Auriculocondylar syndrome 2 caused by a novel PLCB4 variant in a male Chinese neonate: A case report and review of the literature
IF 1.5
4区 医学
Q4 GENETICS & HEREDITY
引用次数: 0
Abstract
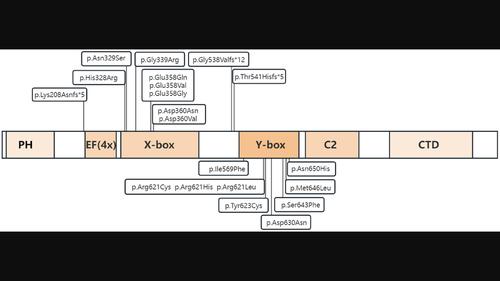
BackgroundAuriculocondylar syndrome (ARCND) is a rare congenital craniofacial developmental malformation syndrome of the first and second pharyngeal arches with external ear malformation at the junction between the lobe and helix, micromaxillary malformation, and mandibular condylar hypoplasia. Four subtypes of ARCND have been described so far, that is, ARCND1 (OMIM # 602483), ARCND2 (ARCND2A, OMIM # 614669; ARCND2B, OMIM # 620458), ARCND3 (OMIM # 615706), and ARCND4 (OMIM # 620457).MethodsThis study reports a case of ARCND2 resulting from a novel pathogenic variant in the
一名中国男性新生儿因新型 PLCB4 变异导致的耳软骨综合征 2:病例报告和文献综述
背景咽喉髁状突综合征(ARCND)是一种罕见的先天性颅面发育畸形综合征,表现为第一和第二咽弓的外耳畸形、耳垂与螺旋交界处的外耳畸形、小颌畸形和下颌骨髁状突发育不良。目前已描述了四种亚型的 ARCND,即 ARCND1(OMIM # 602483)、ARCND2(ARCND2A,OMIM # 614669;ARCND2B,OMIM # 620458)、ARCND3(OMIM # 615706)和 ARCND4(OMIM # 620457)。方法本研究报告了一例由 PLCB4 基因新型致病变异导致的 ARCND2 病例,并总结了 PLCB4 基因的突变位点和 ARCND2 的表型。他患有小颌畸形、小口畸形、独特的问号耳以及下颌骨髁突发育不良。基于三重全外显子组测序确定了 PLCB4 基因中的一个新型错义变异 NM_001377142.1:c.1928C>T(NP_001364071.1:p.Ser643Phe),预计该变异会损害局部结构的稳定性,从而可能影响蛋白质的功能。结论 与其他研究 ARCND2 家族病例的结果一样,在不同家族的 PLCB4 基因杂合突变中观察到不完全的渗透性和不同的表达性。尽管绝大多数 ARCND2 患者的运动和智力发育均处于正常范围,但仍需进行长期随访和评估。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Genetics & Genomic Medicine
Biochemistry, Genetics and Molecular Biology-Genetics
CiteScore
4.20
自引率
0.00%
发文量
241
审稿时长
14 weeks
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


