Further delineation of phenotype and genotype of Kenny–Caffey syndrome type 2 (phenotype and genotype of KCS type 2)
IF 1.6
4区 医学
Q4 GENETICS & HEREDITY
引用次数: 0
Abstract
BackgroundKenny–Caffey syndrome type 2 (KCS2) is an extremely rare inherited disorder characterized by proportionate short stature, skeletal defects, ocular and dental abnormalities, and transient hypocalcemia. It is caused by variants in
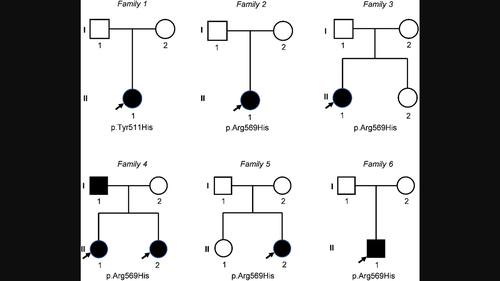
进一步确定肯尼-卡菲综合征 2 型的表型和基因型(肯尼-卡菲综合征 2 型的表型和基因型)
背景肯尼-卡菲综合征 2 型(KCS2)是一种极其罕见的遗传性疾病,以身材矮小、骨骼缺陷、眼部和牙齿异常以及一过性低钙血症为特征。它是由 FAM111A 基因变异引起的。由于 KCS2 与其他综合征相似,缺乏明确的特征,且遗传学确诊病例数量不足,因此诊断 KCS2 具有挑战性。在此,我们旨在进一步描述和总结 KCS2 的基因型和表型,以便更好地了解这种罕见疾病,促进早期诊断和干预。方法我们介绍了来自 6 个家庭的 8 名新患 KCS2 患者的临床和遗传特征,其中一个家庭的 3 名患者被发现是父女相传,为有限的文献资料增添了新的内容。此外,我们还查阅了 PubMed、MEDLINE 和 CNKI 数据库中经基因证实的 KCS2 病例。所有患者均表现为身材矮小(100.0%)。临床表现包括眼部缺陷(5/8)、牙齿问题(3/8)和龋齿(3/8)、骨骼和大脑异常(6/8)、大脑钙化(3/8)、皮质增厚(3/8)和管状骨髓狭窄(4/8)。内分泌异常包括甲状旁腺功能减退(5/8)和低钙血症(3/8)。一名男性患者患有小阴茎和小睾丸症。所有病例均携带FAM111A的错义变异,c.1706核苷酸是突变热点,其中7人携带c.1706G>A(p.Arg569His)变异,1名儿童携带c.1531T>C(p.Tyr511His)变异。文献综述共收集到 20 篇论文中的 46 名患者。数据分析显示,70%以上的KCS2病例存在身材矮小、甲状旁腺功能减退和低钙血症、眼部和牙齿缺陷、骨骼特征(包括皮质增厚和管状骨髓状狭窄)以及癫痫发作/痉挛。我们的研究证实 Arg569His 是热点变异,并总结了 KCS2 的典型表型,这将有助于早期诊断和干预。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Genetics & Genomic Medicine
Biochemistry, Genetics and Molecular Biology-Genetics
CiteScore
4.20
自引率
0.00%
发文量
241
审稿时长
14 weeks
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


