{"title":"Enricherator: A Bayesian Method for Inferring Regularized Genome-wide Enrichments from Sequencing Count Data","authors":"","doi":"10.1016/j.jmb.2024.168567","DOIUrl":null,"url":null,"abstract":"<div><p>A pervasive question in biological research studying gene regulation, chromatin structure, or genomics is where, and to what extent, does a signal of interest arise genome-wide? This question is addressed using a variety of methods relying on high-throughput sequencing data as their final output, including ChIP-seq for protein-DNA interactions,<span><span><sup>1</sup></span></span> GapR-seq for measuring supercoiling,<span><span><sup>2</sup></span></span> and HBD-seq or DRIP-seq for R-loop positioning.<span><span>3</span></span>, <span><span>4</span></span> Current computational methods to calculate genome-wide enrichment of the signal of interest usually do not properly handle the count-based nature of sequencing data, they often do not make use of the local correlation structure of sequencing data, and they do not apply any regularization of enrichment estimates. This can result in unrealistic estimates of the true underlying biological enrichment of interest, unrealistically low estimates of confidence in point estimates of enrichment (or no estimates of confidence at all), unrealistic gyrations in enrichment estimates at very close (<10 bp) genomic loci due to noise inherent in sequencing data, and in a multiple-hypothesis testing problem during interpretation of genome-wide enrichment estimates. We developed a tool called Enricherator to infer genome-wide enrichments from sequencing count data. Enricherator uses the variational Bayes algorithm to fit a generalized linear model to sequencing count data and to sample from the approximate posterior distribution of enrichment estimates (<span><span>https://github.com/jwschroeder3/enricherator</span><svg><path></path></svg></span>). Enrichments inferred by Enricherator more precisely identify known binding sites in cases where low coverage between binding sites leads to false-positive peak calls in these noisy regions of the genome; these benefits extend to published datasets.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 17","pages":"Article 168567"},"PeriodicalIF":4.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022283624001621/pdfft?md5=12eadc9303ecf2b7325490d62b957d44&pid=1-s2.0-S0022283624001621-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624001621","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
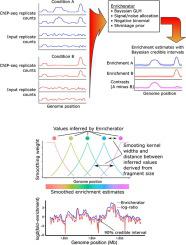
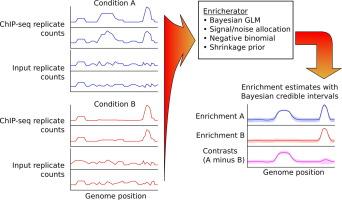
A pervasive question in biological research studying gene regulation, chromatin structure, or genomics is where, and to what extent, does a signal of interest arise genome-wide? This question is addressed using a variety of methods relying on high-throughput sequencing data as their final output, including ChIP-seq for protein-DNA interactions,1 GapR-seq for measuring supercoiling,2 and HBD-seq or DRIP-seq for R-loop positioning.3, 4 Current computational methods to calculate genome-wide enrichment of the signal of interest usually do not properly handle the count-based nature of sequencing data, they often do not make use of the local correlation structure of sequencing data, and they do not apply any regularization of enrichment estimates. This can result in unrealistic estimates of the true underlying biological enrichment of interest, unrealistically low estimates of confidence in point estimates of enrichment (or no estimates of confidence at all), unrealistic gyrations in enrichment estimates at very close (<10 bp) genomic loci due to noise inherent in sequencing data, and in a multiple-hypothesis testing problem during interpretation of genome-wide enrichment estimates. We developed a tool called Enricherator to infer genome-wide enrichments from sequencing count data. Enricherator uses the variational Bayes algorithm to fit a generalized linear model to sequencing count data and to sample from the approximate posterior distribution of enrichment estimates (https://github.com/jwschroeder3/enricherator). Enrichments inferred by Enricherator more precisely identify known binding sites in cases where low coverage between binding sites leads to false-positive peak calls in these noisy regions of the genome; these benefits extend to published datasets.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


