Pierre-François Loos, Antoine Marie and Abdallah Ammar
{"title":"Cumulant Green's function methods for molecules†","authors":"Pierre-François Loos, Antoine Marie and Abdallah Ammar","doi":"10.1039/D4FD00037D","DOIUrl":null,"url":null,"abstract":"<p >The cumulant expansion of the Green's function is a computationally efficient beyond-GW approach renowned for its significant enhancement of satellite features in materials. In contrast to the ubiquitous GW approximation of many-body perturbation theory, <em>ab initio</em> cumulant expansions performed on top of GW (GW + C) have demonstrated the capability to handle multi-particle processes by incorporating higher-order correlation effects or vertex corrections, yielding better agreements between experiment and theory for satellite structures. While widely employed in condensed matter physics, very few applications of GW + C have been published on molecular systems. Here, we assess the performance of this scheme on a series of 10-electron molecular systems (Ne, HF, H<small><sub>2</sub></small>O, NH<small><sub>3</sub></small>, and CH<small><sub>4</sub></small>) where full configuration interaction estimates of the outer-valence quasiparticle and satellite energies are available.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 240-260"},"PeriodicalIF":3.4000,"publicationDate":"2024-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00037d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
Abstract
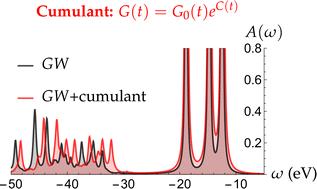
The cumulant expansion of the Green's function is a computationally efficient beyond-GW approach renowned for its significant enhancement of satellite features in materials. In contrast to the ubiquitous GW approximation of many-body perturbation theory, ab initio cumulant expansions performed on top of GW (GW + C) have demonstrated the capability to handle multi-particle processes by incorporating higher-order correlation effects or vertex corrections, yielding better agreements between experiment and theory for satellite structures. While widely employed in condensed matter physics, very few applications of GW + C have been published on molecular systems. Here, we assess the performance of this scheme on a series of 10-electron molecular systems (Ne, HF, H2O, NH3, and CH4) where full configuration interaction estimates of the outer-valence quasiparticle and satellite energies are available.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


