Rima Biswas, Prateek Banerjee, Kavathekar Soham Sudesh
{"title":"Molecular dynamics studies on interfacial interactions between imidazolium-based ionic liquids and carbon nanotubes","authors":"Rima Biswas, Prateek Banerjee, Kavathekar Soham Sudesh","doi":"10.1007/s11224-024-02323-3","DOIUrl":null,"url":null,"abstract":"<div><p>We have investigated the basic mechanism of carbon nanotube (CNT) interactions with various room-temperature ionic liquids (RTILs) using molecular dynamics (MD) simulations. To understand the effects of the cation molecular geometry on the properties of the interface structure in the RTIL systems, we have studied a set of three RTILs with the same [BF<sub>4</sub>]<sup>-</sup> (tetrafluoroborate) anion but with different cations, namely, [EMIM]<sup>+</sup> (1-ethyl-3-methylimidazolium), [BMIM]<sup>+</sup> (1-butyl-3-methylimidazolium), [HMIM]<sup>+</sup> (1-hexyl-3-methylimidazolium), and [OMIM]<sup>+</sup> (1-octyl-3-methylimidazolium) ions. The simulation results showed that the imidazolium cations exhibit two distinct orientations (perpendicular and parallel to the CNTs surface) at the interface irrespective of the alkyl chain length of the cations. The average number of hydrogen bonds per cations inside the CNT was found to be higher for [OMIM][BF<sub>4</sub>] (1.01), which suggests that [OMIM]<sup>+</sup> imidazolium rings to be concentrated at the center of the CNT, which favors hydrogen bond. The reported results show the diffusion coefficients of ions in confinement are much lower in comparison to the bulk region. The interaction energy between [OMIM][BF<sub>4</sub>] (-8.75 kcal.mol<sup>−1</sup>.ion<sup>−1</sup>) and CNT was found to be higher as compared to other ILs. The cations paralleling the CNT surface are thermodynamically significantly more stable because of the substantial interfacial π-π stacking interactions, as shown by a comparison with the calculated interaction energies between cations and the CNTs. Our simulation results provide a molecular-level understanding of the stabilization and dispersion of CNT bundles in ILs.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 6","pages":"1743 - 1753"},"PeriodicalIF":2.1000,"publicationDate":"2024-04-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02323-3","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
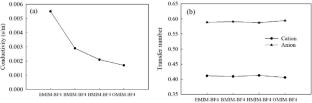
We have investigated the basic mechanism of carbon nanotube (CNT) interactions with various room-temperature ionic liquids (RTILs) using molecular dynamics (MD) simulations. To understand the effects of the cation molecular geometry on the properties of the interface structure in the RTIL systems, we have studied a set of three RTILs with the same [BF4]- (tetrafluoroborate) anion but with different cations, namely, [EMIM]+ (1-ethyl-3-methylimidazolium), [BMIM]+ (1-butyl-3-methylimidazolium), [HMIM]+ (1-hexyl-3-methylimidazolium), and [OMIM]+ (1-octyl-3-methylimidazolium) ions. The simulation results showed that the imidazolium cations exhibit two distinct orientations (perpendicular and parallel to the CNTs surface) at the interface irrespective of the alkyl chain length of the cations. The average number of hydrogen bonds per cations inside the CNT was found to be higher for [OMIM][BF4] (1.01), which suggests that [OMIM]+ imidazolium rings to be concentrated at the center of the CNT, which favors hydrogen bond. The reported results show the diffusion coefficients of ions in confinement are much lower in comparison to the bulk region. The interaction energy between [OMIM][BF4] (-8.75 kcal.mol−1.ion−1) and CNT was found to be higher as compared to other ILs. The cations paralleling the CNT surface are thermodynamically significantly more stable because of the substantial interfacial π-π stacking interactions, as shown by a comparison with the calculated interaction energies between cations and the CNTs. Our simulation results provide a molecular-level understanding of the stabilization and dispersion of CNT bundles in ILs.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


