Geometry-enhanced pretraining on interatomic potentials
IF 18.8
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
Abstract
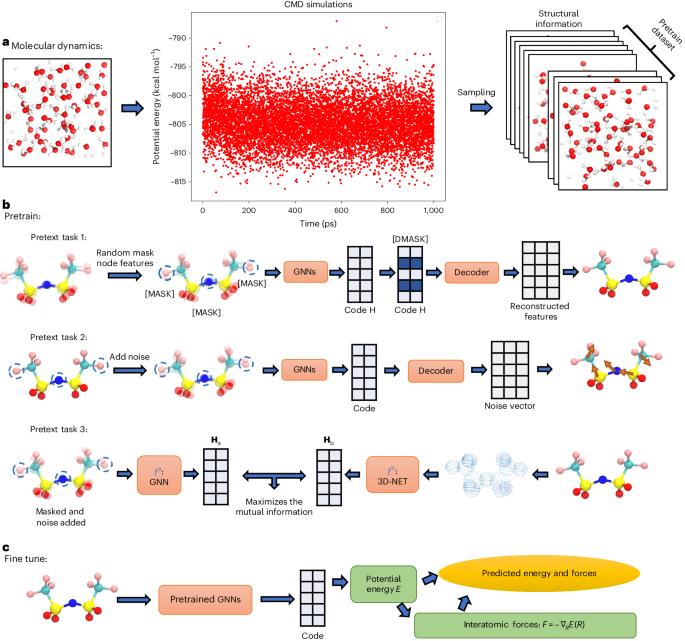
Machine learning interatomic potentials (MLIPs) describe the interactions between atoms in materials and molecules by learning them from a reference database generated by ab initio calculations. MLIPs can accurately and efficiently predict such interactions and have been applied to various fields of physical science. However, high-performance MLIPs rely on a large amount of labelled data, which are costly to obtain by ab initio calculations. Here we propose a geometric structure learning framework that leverages unlabelled configurations to improve the performance of MLIPs. Our framework consists of two stages: first, using classical molecular dynamics simulations to generate unlabelled configurations of the target molecular system; and second, applying geometry-enhanced self-supervised learning techniques, including masking, denoising and contrastive learning, to capture structural information. We evaluate our framework on various benchmarks ranging from small molecule datasets to complex periodic molecular systems with more types of elements. We show that our method significantly improves the accuracy and generalization of MLIPs with only a few additional computational costs and is compatible with different invariant or equivariant graph neural network architectures. Our method enhances MLIPs and advances the simulations of molecular systems. Using machine learning methods to model interatomic potentials enables molecular dynamics simulations with ab initio level accuracy at a relatively low computational cost, but requires a large number of labelled training data obtained through expensive ab initio computations. Cui and colleagues propose a geometric learning framework that leverages self-supervised learning pretraining to enhance existing machine learning based interatomic potential models at a negligible additional computational cost.
几何增强型原子间电位预训练
机器学习原子间势(MLIPs)通过学习由 ab initio 计算生成的参考数据库来描述材料和分子中原子间的相互作用。MLIPs 可以准确、高效地预测这种相互作用,已被应用于物理科学的各个领域。然而,高性能的 MLIPs 依赖于大量的标记数据,而通过 ab initio 计算获得这些数据的成本很高。在这里,我们提出了一个几何结构学习框架,利用未标记的构型来提高 MLIPs 的性能。我们的框架包括两个阶段:首先,利用经典分子动力学模拟生成目标分子系统的无标签构型;其次,应用几何增强型自监督学习技术,包括掩蔽、去噪和对比学习,以捕捉结构信息。我们在从小分子数据集到包含更多元素类型的复杂周期分子系统的各种基准上评估了我们的框架。我们的研究表明,我们的方法大大提高了 MLIPs 的准确性和泛化能力,只增加了少量计算成本,而且与不同的不变或等变图神经网络架构兼容。我们的方法增强了 MLIPs,推进了分子系统的模拟。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


