Lorraina J. Robinson, Eric Goold, David Anderson, Robert C. Rennert, William T. Couldwell, Changhong Xing
{"title":"A mass in the pineal region of a young woman","authors":"Lorraina J. Robinson, Eric Goold, David Anderson, Robert C. Rennert, William T. Couldwell, Changhong Xing","doi":"10.1111/bpa.13258","DOIUrl":null,"url":null,"abstract":"<p>This case involves an 18-year-old female with no significant past medical history who was found to have a large pineal and third ventricular tumor with obstructive hydrocephalus after experiencing subacute, progressive headaches, and an acute episode of sudden vision loss. She underwent a ventriculoperitoneal shunt at an outside facility with improvement in her vision before presenting as a transfer to our facility. Brain magnetic resonance imaging upon transfer demonstrated a large, heterogenous enhancing mass (6.6 × 6.1 × 5.7 cm) centered in the pineal region with extension into the third and right lateral ventricles (Figure 1). The tumor exerted mass effect by splaying the bilateral thalami with effacement of the basal cisterna, effacement of the cerebral aqueduct, and an 8 mm tonsillar herniation. Surgery was pursued for a definitive diagnosis and tumor debulking. Intraoperatively, the tumor demonstrated variable soft and firm consistency with moderate vascularity. Post-operative imaging did not demonstrate clear residual tumor. She was discharged with no new neurologic deficits.</p><p>Histology of H&E-stained sections demonstrated a moderately hypercellular neoplasm consisting of an admixture of small to medium-sized spindle cells embedded in a heavily collagenized and myxoid matrix (Box 1, Figure 2A, B). Tumor cells were observed forming disorganized, haphazard architectural patterns while some tumor cells were seen floating in a loose basophilic myxoid matrix (Figure 2A). The mitotic index was <1/10 high-power-fields. Necrosis was absent. By immunohistochemistry, the tumor cells were patchy positive for CD163 (Figure 2C) and CD99 (Figure 2D), focally positive for desmin (Figure 2D inset), and negative for GFAP, OLIG2, CD34, and SSTR2. MIB-1 proliferation index was overall low (1%–2%) (Figure 2E) but focally increased at approximately 10% (Figure 2E inset). Immunostaining for SMARCB1 showed loss of expression in many of the tumor cells (Figure 2F).</p><p>NeoTYPE brain tumor profile (DNA and RNA) from NeoGenomics was performed, and no pathogenic mutations or fusions were detected. However, a variant of unknown clinical significance involving the <i>SMARCB1</i> gene c.91G>A (p.Glu31Lys) E31K NM_003073.5:c.91G>A was detected. Whole genome methylation profiling was performed at the National Institute of Health (NIH)/National Cancer Institute (NCI) for confirmation of the diagnosis. The composite NIH/NCI methylation profile classified the tumor with a high confidence score (0.99) as a desmoplastic myxoid tumor, <i>SMARCB1</i>-altered.</p><p>Desmoplastic myxoid tumor (DMT) of the pineal region, <i>SMARCB1</i>-mutant.</p><p>As defined by the most recent 2021 World Health Organization (WHO) Central Nervous System tumor classification, DMT of the pineal region, <i>SMARCB1</i>-mutant, maintains a unique <i>SMARCB1</i>-mutation while histologically displaying desmoplasia and myxoid changes but lacking histopathological signs of malignancy. A recent study by Thomas et al. helped to identify the clinical, histopathological, and molecular characterization involving seven cases of this uncommon tumor [<span>1</span>]. Available demographic data from all prior cases [1–3; PMID: 32901946] and the present case indicate a slight female predominance (F: 7; M: 4) and an average age of onset of 34.9 years (range 15–61 years old). All cases have been in the pineal region. Currently, the etiology, pathogenesis, and cell origin of this tumor are unknown. Due to the rarity of this tumor, prognostic and predictive clinical outcomes are still uncertain. Overall, the biological behavior of DMTs with a <i>SMARCB1</i>-mutation appears to be less aggressive than their closely related atypical teratoid/rhabdoid tumor counterparts.</p><p>Histologically, the present case showed the classical morphology observed in this tumor. However, the present case had several features that were not yet described in the previous reports of DMTs. First, the present case had unique immunohistochemical profiles. The present tumor was negative for CD34 and positive for CD163 and CD99, whereas CD34 staining was positive in other reported cases [1–3; PMID: 32901946] and CD163 and CD99 positivity had not been previously reported. The morphology and immunoprofiles of this tumor raise the differential consideration of intracranial mesenchymal tumor, FET::CREB fusion-positive. No fusion was detected on both <i>EWSR1</i> and <i>FUS</i> genes by NeoGenomics brain tumor panel, which excluded this diagnosis. Second, the present case had a novel genetic finding: a missense mutation of <i>SMARCB1</i>, which was not reported in previous studies. In previous reported DMTs [<span>1-3</span>], <i>SMARCB1</i> was mainly inactivated by homozygous or heterozygous deletions or indels rather than point mutations. <i>SMARCB1</i> E31K lies within the DNA-binding region of the SMARCB1 protein (UniProt.org). The reported pathogenic mutations in this amino acid of <i>SMARCB1</i> gene include c.91G>T (p.Glu31Ter) E31* and c.92A>T (p.Glu31Val) E31V. E31K has been identified in sequencing studies (PMID: 33845210, PMID: 37649695), but has not been biochemically characterized, and therefore, its effect on SMARCB1 protein function is unknown.</p><p><i>Clinical data collection</i>: LR, EG, DA, RR, WC, and CX. <i>Figure and manuscript text</i> preparation: LR, RR, EG, DA, and CX. <i>Study supervision</i>: CX. All authors made substantial contributions to the conception or to the acquisition, analysis, or interpretation of data. All authors reviewed and approved the final manuscript.</p><p>The authors have no conflicts of interest to declare.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 3","pages":""},"PeriodicalIF":6.2000,"publicationDate":"2024-03-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13258","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13258","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
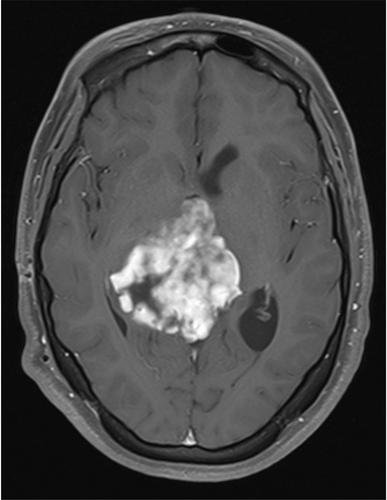
This case involves an 18-year-old female with no significant past medical history who was found to have a large pineal and third ventricular tumor with obstructive hydrocephalus after experiencing subacute, progressive headaches, and an acute episode of sudden vision loss. She underwent a ventriculoperitoneal shunt at an outside facility with improvement in her vision before presenting as a transfer to our facility. Brain magnetic resonance imaging upon transfer demonstrated a large, heterogenous enhancing mass (6.6 × 6.1 × 5.7 cm) centered in the pineal region with extension into the third and right lateral ventricles (Figure 1). The tumor exerted mass effect by splaying the bilateral thalami with effacement of the basal cisterna, effacement of the cerebral aqueduct, and an 8 mm tonsillar herniation. Surgery was pursued for a definitive diagnosis and tumor debulking. Intraoperatively, the tumor demonstrated variable soft and firm consistency with moderate vascularity. Post-operative imaging did not demonstrate clear residual tumor. She was discharged with no new neurologic deficits.
Histology of H&E-stained sections demonstrated a moderately hypercellular neoplasm consisting of an admixture of small to medium-sized spindle cells embedded in a heavily collagenized and myxoid matrix (Box 1, Figure 2A, B). Tumor cells were observed forming disorganized, haphazard architectural patterns while some tumor cells were seen floating in a loose basophilic myxoid matrix (Figure 2A). The mitotic index was <1/10 high-power-fields. Necrosis was absent. By immunohistochemistry, the tumor cells were patchy positive for CD163 (Figure 2C) and CD99 (Figure 2D), focally positive for desmin (Figure 2D inset), and negative for GFAP, OLIG2, CD34, and SSTR2. MIB-1 proliferation index was overall low (1%–2%) (Figure 2E) but focally increased at approximately 10% (Figure 2E inset). Immunostaining for SMARCB1 showed loss of expression in many of the tumor cells (Figure 2F).
NeoTYPE brain tumor profile (DNA and RNA) from NeoGenomics was performed, and no pathogenic mutations or fusions were detected. However, a variant of unknown clinical significance involving the SMARCB1 gene c.91G>A (p.Glu31Lys) E31K NM_003073.5:c.91G>A was detected. Whole genome methylation profiling was performed at the National Institute of Health (NIH)/National Cancer Institute (NCI) for confirmation of the diagnosis. The composite NIH/NCI methylation profile classified the tumor with a high confidence score (0.99) as a desmoplastic myxoid tumor, SMARCB1-altered.
Desmoplastic myxoid tumor (DMT) of the pineal region, SMARCB1-mutant.
As defined by the most recent 2021 World Health Organization (WHO) Central Nervous System tumor classification, DMT of the pineal region, SMARCB1-mutant, maintains a unique SMARCB1-mutation while histologically displaying desmoplasia and myxoid changes but lacking histopathological signs of malignancy. A recent study by Thomas et al. helped to identify the clinical, histopathological, and molecular characterization involving seven cases of this uncommon tumor [1]. Available demographic data from all prior cases [1–3; PMID: 32901946] and the present case indicate a slight female predominance (F: 7; M: 4) and an average age of onset of 34.9 years (range 15–61 years old). All cases have been in the pineal region. Currently, the etiology, pathogenesis, and cell origin of this tumor are unknown. Due to the rarity of this tumor, prognostic and predictive clinical outcomes are still uncertain. Overall, the biological behavior of DMTs with a SMARCB1-mutation appears to be less aggressive than their closely related atypical teratoid/rhabdoid tumor counterparts.
Histologically, the present case showed the classical morphology observed in this tumor. However, the present case had several features that were not yet described in the previous reports of DMTs. First, the present case had unique immunohistochemical profiles. The present tumor was negative for CD34 and positive for CD163 and CD99, whereas CD34 staining was positive in other reported cases [1–3; PMID: 32901946] and CD163 and CD99 positivity had not been previously reported. The morphology and immunoprofiles of this tumor raise the differential consideration of intracranial mesenchymal tumor, FET::CREB fusion-positive. No fusion was detected on both EWSR1 and FUS genes by NeoGenomics brain tumor panel, which excluded this diagnosis. Second, the present case had a novel genetic finding: a missense mutation of SMARCB1, which was not reported in previous studies. In previous reported DMTs [1-3], SMARCB1 was mainly inactivated by homozygous or heterozygous deletions or indels rather than point mutations. SMARCB1 E31K lies within the DNA-binding region of the SMARCB1 protein (UniProt.org). The reported pathogenic mutations in this amino acid of SMARCB1 gene include c.91G>T (p.Glu31Ter) E31* and c.92A>T (p.Glu31Val) E31V. E31K has been identified in sequencing studies (PMID: 33845210, PMID: 37649695), but has not been biochemically characterized, and therefore, its effect on SMARCB1 protein function is unknown.
Clinical data collection: LR, EG, DA, RR, WC, and CX. Figure and manuscript text preparation: LR, RR, EG, DA, and CX. Study supervision: CX. All authors made substantial contributions to the conception or to the acquisition, analysis, or interpretation of data. All authors reviewed and approved the final manuscript.
The authors have no conflicts of interest to declare.
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


