{"title":"DockThor-VS: A Free Platform for Receptor-Ligand Virtual Screening","authors":"","doi":"10.1016/j.jmb.2024.168548","DOIUrl":null,"url":null,"abstract":"<div><p>The DockThor-VS platform (<span><span>https://dockthor.lncc.br/v2/</span><svg><path></path></svg></span>) is a free protein–ligand docking server conceptualized to facilitate and assist drug discovery projects to perform docking-based virtual screening experiments accurately and using high-performance computing. The DockThor docking engine is a grid-based method designed for flexible-ligand and rigid-receptor docking. It employs a multiple-solution genetic algorithm and the MMFF94S molecular force field scoring function for pose prediction. This engine was engineered to handle highly flexible ligands, such as peptides. Affinity prediction and ranking of protein–ligand complexes are performed with the linear empirical scoring function DockTScore. The main steps of the ligand and protein preparation are available on the DockThor Portal, making it possible to change the protonation states of the amino acid residues, and include cofactors as rigid entities. The user can also customize and visualize the main parameters of the grid box. The results of docking experiments are automatically clustered and ordered, providing users with a diverse array of meaningful binding modes. The platform DockThor-VS offers a user-friendly interface and powerful algorithms, enabling researchers to conduct virtual screening experiments efficiently and accurately. The DockThor Portal utilizes the computational strength of the Brazilian high-performance platform SDumont, further amplifying the efficiency and speed of docking experiments. Additionally, the web server facilitates and enhances virtual screening experiments by offering curated structures of potential targets and compound datasets, such as proteins related to COVID-19 and FDA-approved drugs for repurposing studies. In summary, DockThor-VS is a dynamic and evolving solution for docking-based virtual screening to be applied in drug discovery projects.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 17","pages":"Article 168548"},"PeriodicalIF":4.5000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022283624001438/pdfft?md5=57349e8fba1907bce8b7894215619c89&pid=1-s2.0-S0022283624001438-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624001438","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
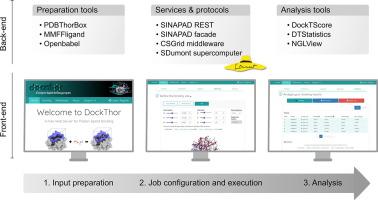
The DockThor-VS platform (https://dockthor.lncc.br/v2/) is a free protein–ligand docking server conceptualized to facilitate and assist drug discovery projects to perform docking-based virtual screening experiments accurately and using high-performance computing. The DockThor docking engine is a grid-based method designed for flexible-ligand and rigid-receptor docking. It employs a multiple-solution genetic algorithm and the MMFF94S molecular force field scoring function for pose prediction. This engine was engineered to handle highly flexible ligands, such as peptides. Affinity prediction and ranking of protein–ligand complexes are performed with the linear empirical scoring function DockTScore. The main steps of the ligand and protein preparation are available on the DockThor Portal, making it possible to change the protonation states of the amino acid residues, and include cofactors as rigid entities. The user can also customize and visualize the main parameters of the grid box. The results of docking experiments are automatically clustered and ordered, providing users with a diverse array of meaningful binding modes. The platform DockThor-VS offers a user-friendly interface and powerful algorithms, enabling researchers to conduct virtual screening experiments efficiently and accurately. The DockThor Portal utilizes the computational strength of the Brazilian high-performance platform SDumont, further amplifying the efficiency and speed of docking experiments. Additionally, the web server facilitates and enhances virtual screening experiments by offering curated structures of potential targets and compound datasets, such as proteins related to COVID-19 and FDA-approved drugs for repurposing studies. In summary, DockThor-VS is a dynamic and evolving solution for docking-based virtual screening to be applied in drug discovery projects.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


