Yingdong Song, Tao Shen, Huihui Sun, Xiangting Wang
{"title":"Genome-wide analyses reveal the regulatory roles of DNA methylation-regulated alternative promoter transcripts in breast cancer","authors":"Yingdong Song, Tao Shen, Huihui Sun, Xiangting Wang","doi":"10.1007/s00439-024-02653-6","DOIUrl":null,"url":null,"abstract":"<p>A certain proportion of genes are regulated by multiple, distinct promoters, revealing a dynamic landscape of the cancer transcriptome. However, the contribution of alternative promoters (APs) in breast cancer (BRCA) remains largely unexplored. Here, we identified 3654 genes with multiple promoters in BRCA patients, and 53 of them could generate distinct AP transcripts that are dysregulated and prognosis-related in BRCA, namely prognosis-related dysregulated AP (prdeAP) transcripts. Interestingly, when we searched for the genomic signatures of these prdeAP genes, we found that the promoter regions of 92% of the prdeAP genes were enriched with abundant DNA methylation signals. Through further bioinformatic analysis and experimental validation, we showed that AP selections of <i>TANK</i>, <i>UNKL</i>, <i>CCL28</i>, and <i>MAP1LC3A</i> were regulated by DNA methylation upon their corresponding promoter regions. Functionally, by overexpressing AP variants of <i>TANK</i>, we found that <i>TANK|55731</i> could dramatically suppress MDA-MB-231 cell proliferation and migration. Meanwhile, pan-cancer survival analyses suggested that AP variants of <i>TANK</i> provided more accurate prognostic predictive ability than <i>TANK</i> gene in a variety of tumor types, including BRCA. Together, by uncovering the DNA methylation-regulated AP transcripts with tumor prognostic features, our work revealed a novel layer of regulators in BRCA progression and provided potential targets that served as effective biomarkers for anti-BRCA treatment.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":"26 1","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02653-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
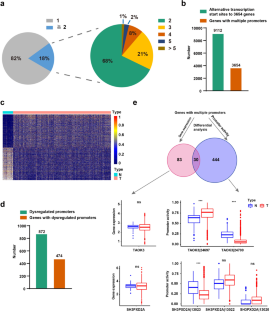
A certain proportion of genes are regulated by multiple, distinct promoters, revealing a dynamic landscape of the cancer transcriptome. However, the contribution of alternative promoters (APs) in breast cancer (BRCA) remains largely unexplored. Here, we identified 3654 genes with multiple promoters in BRCA patients, and 53 of them could generate distinct AP transcripts that are dysregulated and prognosis-related in BRCA, namely prognosis-related dysregulated AP (prdeAP) transcripts. Interestingly, when we searched for the genomic signatures of these prdeAP genes, we found that the promoter regions of 92% of the prdeAP genes were enriched with abundant DNA methylation signals. Through further bioinformatic analysis and experimental validation, we showed that AP selections of TANK, UNKL, CCL28, and MAP1LC3A were regulated by DNA methylation upon their corresponding promoter regions. Functionally, by overexpressing AP variants of TANK, we found that TANK|55731 could dramatically suppress MDA-MB-231 cell proliferation and migration. Meanwhile, pan-cancer survival analyses suggested that AP variants of TANK provided more accurate prognostic predictive ability than TANK gene in a variety of tumor types, including BRCA. Together, by uncovering the DNA methylation-regulated AP transcripts with tumor prognostic features, our work revealed a novel layer of regulators in BRCA progression and provided potential targets that served as effective biomarkers for anti-BRCA treatment.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


