{"title":"Molecular insights into the binding interactions and energetics of the omicron spike variant with hACE2 and a neutralizing antibody","authors":"Vipul Kumar , Seyad Shefrin, Durai Sundar","doi":"10.1016/j.jsb.2024.108087","DOIUrl":null,"url":null,"abstract":"<div><p>The global spread of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) since 2019 has led to a continuous evolution of viral variants, with the latest concern being the Omicron (B.1.1.529) variant. In this study, classical molecular dynamics simulations were conducted to elucidate the biophysical aspects of the Omicron spike protein's receptor-binding domain (RBD) in its interaction with human angiotensin-converting enzyme 2 (hACE2) and a neutralizing antibody, comparing it to the wildtype (WT). To model the Omicron variant, 15 in silico mutations were introduced in the RBD region of WT (retrieved from PDB). The simulations of WT spike-hACE2 and Omicron spike-hACE2 complexes revealed comparable binding stability and dynamics. Notably, the Q493R mutation in the Omicron spike increased interactions with hACE2, particularly with ASP38 and ASP355. Additionally, mutations such as N417K, T478K, and Y505H contributed to enhanced structural stability in the Omicron variant. Conversely, when comparing WT with Omicron in complex with a neutralizing antibody, simulation results demonstrated poorer binding dynamics and stability for the Omicron variant. The E484K mutation significantly decreased binding interactions, resulting in an overall decrease in binding energy (∼−57 kcal/mol) compared to WT (∼−84 kcal/mol). This study provides valuable molecular insights into the heightened infectivity of the Omicron variant, shedding light on the specific mutations influencing its interactions with hACE2 and neutralizing antibodies.</p></div>","PeriodicalId":17074,"journal":{"name":"Journal of structural biology","volume":"216 2","pages":"Article 108087"},"PeriodicalIF":2.7000,"publicationDate":"2024-03-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of structural biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1047847724000273","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
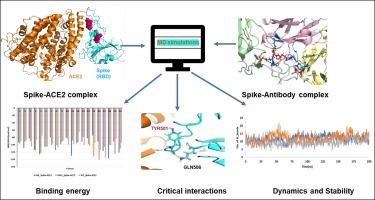
The global spread of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) since 2019 has led to a continuous evolution of viral variants, with the latest concern being the Omicron (B.1.1.529) variant. In this study, classical molecular dynamics simulations were conducted to elucidate the biophysical aspects of the Omicron spike protein's receptor-binding domain (RBD) in its interaction with human angiotensin-converting enzyme 2 (hACE2) and a neutralizing antibody, comparing it to the wildtype (WT). To model the Omicron variant, 15 in silico mutations were introduced in the RBD region of WT (retrieved from PDB). The simulations of WT spike-hACE2 and Omicron spike-hACE2 complexes revealed comparable binding stability and dynamics. Notably, the Q493R mutation in the Omicron spike increased interactions with hACE2, particularly with ASP38 and ASP355. Additionally, mutations such as N417K, T478K, and Y505H contributed to enhanced structural stability in the Omicron variant. Conversely, when comparing WT with Omicron in complex with a neutralizing antibody, simulation results demonstrated poorer binding dynamics and stability for the Omicron variant. The E484K mutation significantly decreased binding interactions, resulting in an overall decrease in binding energy (∼−57 kcal/mol) compared to WT (∼−84 kcal/mol). This study provides valuable molecular insights into the heightened infectivity of the Omicron variant, shedding light on the specific mutations influencing its interactions with hACE2 and neutralizing antibodies.
期刊介绍:
Journal of Structural Biology (JSB) has an open access mirror journal, the Journal of Structural Biology: X (JSBX), sharing the same aims and scope, editorial team, submission system and rigorous peer review. Since both journals share the same editorial system, you may submit your manuscript via either journal homepage. You will be prompted during submission (and revision) to choose in which to publish your article. The editors and reviewers are not aware of the choice you made until the article has been published online. JSB and JSBX publish papers dealing with the structural analysis of living material at every level of organization by all methods that lead to an understanding of biological function in terms of molecular and supermolecular structure.
Techniques covered include:
• Light microscopy including confocal microscopy
• All types of electron microscopy
• X-ray diffraction
• Nuclear magnetic resonance
• Scanning force microscopy, scanning probe microscopy, and tunneling microscopy
• Digital image processing
• Computational insights into structure

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


