B. P. Norman, H. Sutherland, P. J. M. Wilson, D. A. Rutland, A. M. Milan, A. T. Hughes, A. S. Davison, M. Khedr, J. C. Jarvis, J. A. Gallagher, G. Bou-Gharios, L. R. Ranganath
{"title":"Hepatobiliary circulation and dominant urinary excretion of homogentisic acid in a mouse model of alkaptonuria","authors":"B. P. Norman, H. Sutherland, P. J. M. Wilson, D. A. Rutland, A. M. Milan, A. T. Hughes, A. S. Davison, M. Khedr, J. C. Jarvis, J. A. Gallagher, G. Bou-Gharios, L. R. Ranganath","doi":"10.1002/jimd.12728","DOIUrl":null,"url":null,"abstract":"<p>Altered activity of specific enzymes in phenylalanine-tyrosine (phe-tyr) metabolism results in incomplete breakdown of various metabolite substrates in this pathway. Increased biofluid concentration and tissue accumulation of the phe-tyr pathway metabolite homogentisic acid (HGA) is central to pathophysiology in the inherited disorder alkaptonuria (AKU). Accumulation of metabolites upstream of HGA, including tyrosine, occurs in patients on nitisinone, a licenced drug for AKU and hereditary tyrosinaemia type 1, which inhibits the enzyme responsible for HGA production. The aim of this study was to investigate the phe-tyr metabolite content of key biofluids and tissues in AKU mice on and off nitisinone to gain new insights into the biodistribution of metabolites in these altered metabolic states. The data show for the first time that HGA is present in bile in AKU (mean [±SD] = 1003[±410] μmol/L; nitisinone-treated AKU mean [±SD] = 45[±23] μmol/L). Biliary tyrosine, 3(4-hydroxyphenyl)pyruvic acid (HPPA) and 3(4-hydroxyphenyl)lactic acid (HPLA) are also increased on nitisinone. Urine was confirmed as the dominant elimination route of HGA in untreated AKU, but with indication of biliary excretion. These data provide new insights into pathways of phe-tyr metabolite biodistribution and metabolism, showing for the first time that hepatobiliary excretion contributes to the total pool of metabolites in this pathway. Our data suggest that biliary elimination of organic acids and other metabolites may play an underappreciated role in disorders of metabolism. We propose that our finding of approximately 3.8 times greater urinary HGA excretion in AKU mice compared with patients is one reason for the lack of extensive tissue ochronosis in the AKU mouse model.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 4","pages":"664-673"},"PeriodicalIF":4.2000,"publicationDate":"2024-03-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12728","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12728","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
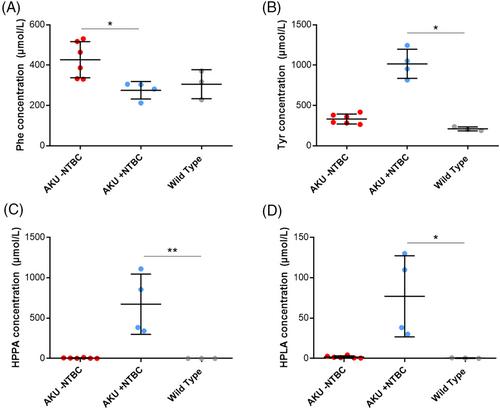
Altered activity of specific enzymes in phenylalanine-tyrosine (phe-tyr) metabolism results in incomplete breakdown of various metabolite substrates in this pathway. Increased biofluid concentration and tissue accumulation of the phe-tyr pathway metabolite homogentisic acid (HGA) is central to pathophysiology in the inherited disorder alkaptonuria (AKU). Accumulation of metabolites upstream of HGA, including tyrosine, occurs in patients on nitisinone, a licenced drug for AKU and hereditary tyrosinaemia type 1, which inhibits the enzyme responsible for HGA production. The aim of this study was to investigate the phe-tyr metabolite content of key biofluids and tissues in AKU mice on and off nitisinone to gain new insights into the biodistribution of metabolites in these altered metabolic states. The data show for the first time that HGA is present in bile in AKU (mean [±SD] = 1003[±410] μmol/L; nitisinone-treated AKU mean [±SD] = 45[±23] μmol/L). Biliary tyrosine, 3(4-hydroxyphenyl)pyruvic acid (HPPA) and 3(4-hydroxyphenyl)lactic acid (HPLA) are also increased on nitisinone. Urine was confirmed as the dominant elimination route of HGA in untreated AKU, but with indication of biliary excretion. These data provide new insights into pathways of phe-tyr metabolite biodistribution and metabolism, showing for the first time that hepatobiliary excretion contributes to the total pool of metabolites in this pathway. Our data suggest that biliary elimination of organic acids and other metabolites may play an underappreciated role in disorders of metabolism. We propose that our finding of approximately 3.8 times greater urinary HGA excretion in AKU mice compared with patients is one reason for the lack of extensive tissue ochronosis in the AKU mouse model.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


