A Paripović, A Maver, N Stajić, J Putnik, S Ostojić, B Alimpić, N Ilić, A Sarajlija
{"title":"Expanding the Phenotypic Spectrum: Chronic Kidney Disease in a Patient with Combined Oxidative Phosphorylation Defect 21.","authors":"A Paripović, A Maver, N Stajić, J Putnik, S Ostojić, B Alimpić, N Ilić, A Sarajlija","doi":"10.2478/bjmg-2023-0016","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Pathogenic variants in <i>TARS2</i> are associated with combined oxidative phosphorylation deficiency 21 (COXPD21), an autosomal recessive disorder usually presenting as mitochondrial encephalomyopathy. Kidney impairment has been documented in a minority of COXPD21 patients, mostly with distal renal tubular acidosis.</p><p><strong>Case report: </strong>We report on the first COXPD21 patient with generalized tubular dysfunction and early childhood progression to chronic kidney disease (CKD). Thorough diagnostic evaluation was initiated at six months of age due to failure to thrive, muscular hypotonia, motor delay and recurrent bronchiolitis. The boy was lost to follow-up until the age of two years, when he was readmitted with elevated creatinine level, reduced estimated glomerular filtrate rate, normochromic anaemia, metabolic acidosis and hyperkalaemia. Urine abnormalities pointed to generalized tubular dysfunction. Two novel heterozygous missense variants in <i>TARS2</i> gene were detected by the means of whole exome sequencing: c.1298T>G (p.Phe438Cys) of maternal origin and c.1931A>T (p.Asp644Val) of paternal origin. Currently, at 4.5 years of age, the boy has failure to thrive, severe motor and verbal delay and end stage of CKD. We referred the patient to paediatric centre that provides renal replacement therapy.</p><p><strong>Conclusion: </strong>The overall clinical course in the patient we report on corresponds well to the previously reported cases of <i>TARS2</i> related COXPD21, especially in regard to neurological and developmental aspects of the disease. However, we point out the generalized tubulopathy and early occurrence of CKD in our patient as atypical renal involvement in COXPD21. Additionally, this is the first report of hypothyroidism and hypoparathyroidism in a COXPD21 patient.</p>","PeriodicalId":55403,"journal":{"name":"Balkan Journal of Medical Genetics","volume":"26 2","pages":"59-64"},"PeriodicalIF":0.9000,"publicationDate":"2024-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10932596/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Balkan Journal of Medical Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2478/bjmg-2023-0016","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Pathogenic variants in TARS2 are associated with combined oxidative phosphorylation deficiency 21 (COXPD21), an autosomal recessive disorder usually presenting as mitochondrial encephalomyopathy. Kidney impairment has been documented in a minority of COXPD21 patients, mostly with distal renal tubular acidosis.
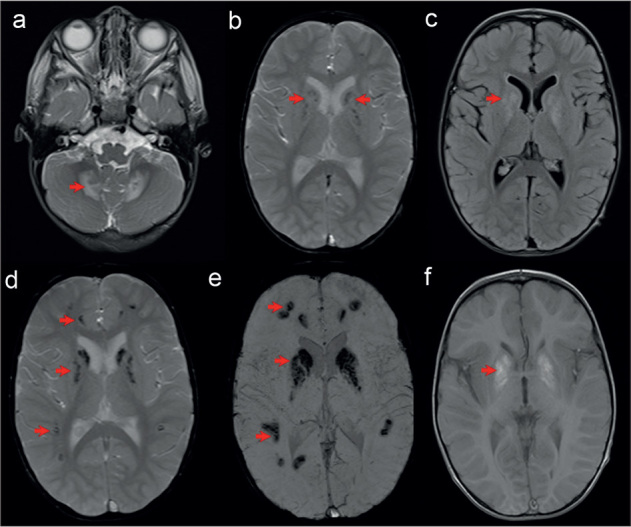
Case report: We report on the first COXPD21 patient with generalized tubular dysfunction and early childhood progression to chronic kidney disease (CKD). Thorough diagnostic evaluation was initiated at six months of age due to failure to thrive, muscular hypotonia, motor delay and recurrent bronchiolitis. The boy was lost to follow-up until the age of two years, when he was readmitted with elevated creatinine level, reduced estimated glomerular filtrate rate, normochromic anaemia, metabolic acidosis and hyperkalaemia. Urine abnormalities pointed to generalized tubular dysfunction. Two novel heterozygous missense variants in TARS2 gene were detected by the means of whole exome sequencing: c.1298T>G (p.Phe438Cys) of maternal origin and c.1931A>T (p.Asp644Val) of paternal origin. Currently, at 4.5 years of age, the boy has failure to thrive, severe motor and verbal delay and end stage of CKD. We referred the patient to paediatric centre that provides renal replacement therapy.
Conclusion: The overall clinical course in the patient we report on corresponds well to the previously reported cases of TARS2 related COXPD21, especially in regard to neurological and developmental aspects of the disease. However, we point out the generalized tubulopathy and early occurrence of CKD in our patient as atypical renal involvement in COXPD21. Additionally, this is the first report of hypothyroidism and hypoparathyroidism in a COXPD21 patient.
期刊介绍:
Balkan Journal of Medical Genetics is a journal in the English language for publication of articles involving all branches of medical genetics: human cytogenetics, molecular genetics, clinical genetics, immunogenetics, oncogenetics, pharmacogenetics, population genetics, genetic screening and diagnosis of monogenic and polygenic diseases, prenatal and preimplantation genetic diagnosis, genetic counselling, advances in treatment and prevention.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


