{"title":"Crystal Structure Assignment for Unknown Compounds from X-ray Diffraction Patterns with Deep Learning","authors":"Litao Chen, Bingxu Wang, Wentao Zhang, Shisheng Zheng, Zhefeng Chen, Mingzheng Zhang, Cheng Dong, Feng Pan* and Shunning Li*, ","doi":"10.1021/jacs.3c11852","DOIUrl":null,"url":null,"abstract":"<p >Determining the structures of previously unseen compounds from experimental characterizations is a crucial part of materials science. It requires a step of searching for the structure type that conforms to the lattice of the unknown compound, which enables the pattern matching process for characterization data, such as X-ray diffraction (XRD) patterns. However, this procedure typically places a high demand on domain expertise, thus creating an obstacle for computer-driven automation. Here, we address this challenge by leveraging a deep-learning model composed of a union of convolutional residual neural networks. The accuracy of the model is demonstrated on a dataset of over 60,000 different compounds for 100 structure types, and additional categories can be integrated without the need to retrain the existing networks. We also unravel the operation of the deep-learning black box and highlight the way in which the resemblance between the unknown compound and a structure type is quantified based on both local and global characteristics in XRD patterns. This computational tool opens new avenues for automating structure analysis on materials unearthed in high-throughput experimentation.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":"146 12","pages":"8098–8109"},"PeriodicalIF":14.4000,"publicationDate":"2024-03-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jacs.3c11852","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
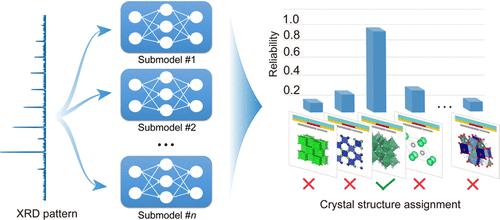
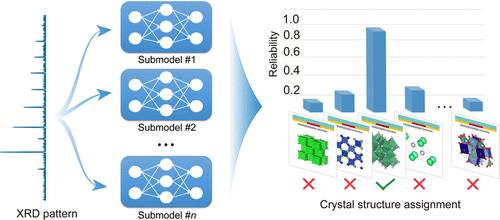
Determining the structures of previously unseen compounds from experimental characterizations is a crucial part of materials science. It requires a step of searching for the structure type that conforms to the lattice of the unknown compound, which enables the pattern matching process for characterization data, such as X-ray diffraction (XRD) patterns. However, this procedure typically places a high demand on domain expertise, thus creating an obstacle for computer-driven automation. Here, we address this challenge by leveraging a deep-learning model composed of a union of convolutional residual neural networks. The accuracy of the model is demonstrated on a dataset of over 60,000 different compounds for 100 structure types, and additional categories can be integrated without the need to retrain the existing networks. We also unravel the operation of the deep-learning black box and highlight the way in which the resemblance between the unknown compound and a structure type is quantified based on both local and global characteristics in XRD patterns. This computational tool opens new avenues for automating structure analysis on materials unearthed in high-throughput experimentation.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


