Felix Zeller, Chieh-Min Hsieh, Wilke Dononelli, Tim Neudecker
{"title":"Computational high-pressure chemistry: Ab initio simulations of atoms, molecules, and extended materials in the gigapascal regime","authors":"Felix Zeller, Chieh-Min Hsieh, Wilke Dononelli, Tim Neudecker","doi":"10.1002/wcms.1708","DOIUrl":null,"url":null,"abstract":"<p>The field of liquid-phase and solid-state high-pressure chemistry has exploded since the advent of the diamond anvil cell, an experimental technique that allows the application of pressures up to several hundred gigapascals. To complement high-pressure experiments, a large number of computational tools have been developed. These techniques enable the simulation of chemical systems, their sizes ranging from single atoms to infinitely large crystals, under high pressure, and the calculation of the resulting structural, electronic, and spectroscopic changes. At the most fundamental level, computational methods using carefully tailored wall potentials allow the analytical calculation of energies and electronic properties of compressed atoms. Molecules and molecular clusters can be compressed either via mechanochemical approaches or via more sophisticated computational protocols using implicit or explicit solvation approaches, typically in combination with density functional theory, thus allowing the simulation of pressure-induced chemical reactions. Crystals and other periodic systems can be routinely simulated under pressure as well, both statically and dynamically, to predict the changes of crystallographic data under pressure and high-pressure crystal structure transitions. In this review, the theoretical foundations of the available computational tools for simulating high-pressure chemistry are introduced and example applications demonstrating the strengths and weaknesses of each approach are discussed.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 2","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2024-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1708","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1708","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
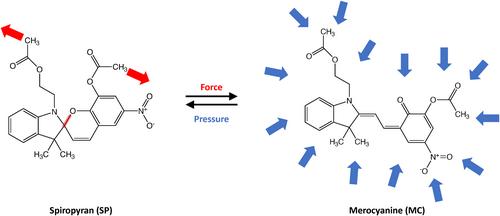
The field of liquid-phase and solid-state high-pressure chemistry has exploded since the advent of the diamond anvil cell, an experimental technique that allows the application of pressures up to several hundred gigapascals. To complement high-pressure experiments, a large number of computational tools have been developed. These techniques enable the simulation of chemical systems, their sizes ranging from single atoms to infinitely large crystals, under high pressure, and the calculation of the resulting structural, electronic, and spectroscopic changes. At the most fundamental level, computational methods using carefully tailored wall potentials allow the analytical calculation of energies and electronic properties of compressed atoms. Molecules and molecular clusters can be compressed either via mechanochemical approaches or via more sophisticated computational protocols using implicit or explicit solvation approaches, typically in combination with density functional theory, thus allowing the simulation of pressure-induced chemical reactions. Crystals and other periodic systems can be routinely simulated under pressure as well, both statically and dynamically, to predict the changes of crystallographic data under pressure and high-pressure crystal structure transitions. In this review, the theoretical foundations of the available computational tools for simulating high-pressure chemistry are introduced and example applications demonstrating the strengths and weaknesses of each approach are discussed.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


