Jianfeng Wang , Jinling Liu , Jingjing Shao , Hongyu Chen , Luyun Cui , Pei Zhang , Yinan Yao , Jianying Zhou , Zhang Bao
{"title":"Cigarette smoking inhibits myoblast regeneration by promoting proteasomal degradation of NPAT protein and hindering cell cycle progression","authors":"Jianfeng Wang , Jinling Liu , Jingjing Shao , Hongyu Chen , Luyun Cui , Pei Zhang , Yinan Yao , Jianying Zhou , Zhang Bao","doi":"10.1016/j.crtox.2024.100161","DOIUrl":null,"url":null,"abstract":"<div><p>Cigarette smoking (CS) causes skeletal muscle dysfunction, leading to sarcopenia and worse prognosis of patients with diverse systemic diseases. Here, we found that CS exposure prevented C2C12 myoblasts proliferation in a dose-dependent manner. Immunoblotting assays verified that CS exposure promoted the expression of cell cycle suppressor protein p21. Furthermore, CS exposure significantly inhibited replication-dependent (RD) histone transcription and caused S phase arrest in the cell cycle during C2C12 proliferation. Mechanistically, CS deregulated the expression levels of Nuclear Protein Ataxia-Telangiectasia Locus (NPAT/p220). Notably, the proteasome inhibitor MG132 was able to reverse the expression of NPAT in myoblasts, implying that the degradation of CS-mediated NPAT is proteasome-dependent. Overexpression of NPAT also rescued the defective proliferation phenotype induced by CS in C2C12 myoblasts. Taken together, we suggest that CS exposure induces NPAT degradation in C2C12 myoblasts and impairs myogenic proliferation through NPAT associated proteasomal-dependent mechanisms. As an application of the proteasome inhibitor MG132 or overexpression of NPAT could reverse the impaired proliferation of myoblasts induced by CS, the recovery of myoblast proliferation may be potential strategies to treat CS-related skeletal muscle dysfunction.</p></div>","PeriodicalId":11236,"journal":{"name":"Current Research in Toxicology","volume":"6 ","pages":"Article 100161"},"PeriodicalIF":2.9000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2666027X24000148/pdfft?md5=62b76c5eac046a7203d59ddece106329&pid=1-s2.0-S2666027X24000148-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Research in Toxicology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666027X24000148","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"TOXICOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
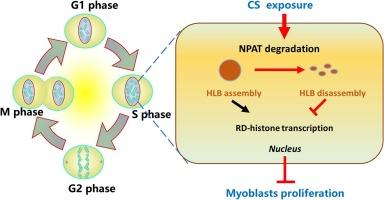
Cigarette smoking (CS) causes skeletal muscle dysfunction, leading to sarcopenia and worse prognosis of patients with diverse systemic diseases. Here, we found that CS exposure prevented C2C12 myoblasts proliferation in a dose-dependent manner. Immunoblotting assays verified that CS exposure promoted the expression of cell cycle suppressor protein p21. Furthermore, CS exposure significantly inhibited replication-dependent (RD) histone transcription and caused S phase arrest in the cell cycle during C2C12 proliferation. Mechanistically, CS deregulated the expression levels of Nuclear Protein Ataxia-Telangiectasia Locus (NPAT/p220). Notably, the proteasome inhibitor MG132 was able to reverse the expression of NPAT in myoblasts, implying that the degradation of CS-mediated NPAT is proteasome-dependent. Overexpression of NPAT also rescued the defective proliferation phenotype induced by CS in C2C12 myoblasts. Taken together, we suggest that CS exposure induces NPAT degradation in C2C12 myoblasts and impairs myogenic proliferation through NPAT associated proteasomal-dependent mechanisms. As an application of the proteasome inhibitor MG132 or overexpression of NPAT could reverse the impaired proliferation of myoblasts induced by CS, the recovery of myoblast proliferation may be potential strategies to treat CS-related skeletal muscle dysfunction.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


