{"title":"Integration of graph neural networks and genome-scale metabolic models for predicting gene essentiality","authors":"Ramin Hasibi, Tom Michoel, Diego A. Oyarzún","doi":"10.1038/s41540-024-00348-2","DOIUrl":null,"url":null,"abstract":"<p>Genome-scale metabolic models are powerful tools for understanding cellular physiology. Flux balance analysis (FBA), in particular, is an optimization-based approach widely employed for predicting metabolic phenotypes. In model microbes such as <i>Escherichia coli</i>, FBA has been successful at predicting essential genes, i.e. those genes that impair survival when deleted. A central assumption in this approach is that both wild type and deletion strains optimize the same fitness objective. Although the optimality assumption may hold for the wild type metabolic network, deletion strains are not subject to the same evolutionary pressures and knock-out mutants may steer their metabolism to meet other objectives for survival. Here, we present FlowGAT, a hybrid FBA-machine learning strategy for predicting essentiality directly from wild type metabolic phenotypes. The approach is based on graph-structured representation of metabolic fluxes predicted by FBA, where nodes correspond to enzymatic reactions and edges quantify the propagation of metabolite mass flow between a reaction and its neighbours. We integrate this information into a graph neural network that can be trained on knock-out fitness assay data. Comparisons across different model architectures reveal that FlowGAT predictions for <i>E. coli</i> are close to those of FBA for several growth conditions. This suggests that essentiality of enzymatic genes can be predicted by exploiting the inherent network structure of metabolism. Our approach demonstrates the benefits of combining the mechanistic insights afforded by genome-scale models with the ability of deep learning to infer patterns from complex datasets.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"2 1","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2024-03-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00348-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
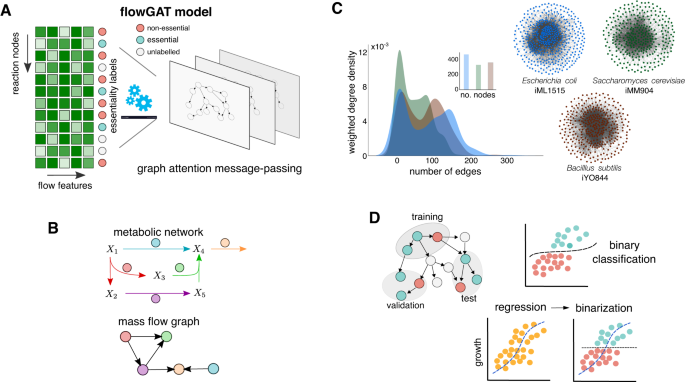
Genome-scale metabolic models are powerful tools for understanding cellular physiology. Flux balance analysis (FBA), in particular, is an optimization-based approach widely employed for predicting metabolic phenotypes. In model microbes such as Escherichia coli, FBA has been successful at predicting essential genes, i.e. those genes that impair survival when deleted. A central assumption in this approach is that both wild type and deletion strains optimize the same fitness objective. Although the optimality assumption may hold for the wild type metabolic network, deletion strains are not subject to the same evolutionary pressures and knock-out mutants may steer their metabolism to meet other objectives for survival. Here, we present FlowGAT, a hybrid FBA-machine learning strategy for predicting essentiality directly from wild type metabolic phenotypes. The approach is based on graph-structured representation of metabolic fluxes predicted by FBA, where nodes correspond to enzymatic reactions and edges quantify the propagation of metabolite mass flow between a reaction and its neighbours. We integrate this information into a graph neural network that can be trained on knock-out fitness assay data. Comparisons across different model architectures reveal that FlowGAT predictions for E. coli are close to those of FBA for several growth conditions. This suggests that essentiality of enzymatic genes can be predicted by exploiting the inherent network structure of metabolism. Our approach demonstrates the benefits of combining the mechanistic insights afforded by genome-scale models with the ability of deep learning to infer patterns from complex datasets.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


