{"title":"Okur-Chung neurodevelopmental syndrome: Implications for phenotype and genotype expansion.","authors":"Haitian Nan, Min Chu, Jing Zhang, Deming Jiang, Yihao Wang, Liyong Wu","doi":"10.1002/mgg3.2398","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Okur-Chung neurodevelopmental syndrome (OCNDS) is a rare autosomal dominant disorder caused by pathogenic variants in CSNK2A1. It is characterized by intellectual disability, developmental delay, and multisystemic abnormalities.</p><p><strong>Methods: </strong>We performed the whole-exome sequencing for a patient in a Chinese family. The co-segregation study using the Sanger sequencing method was performed among family members. Reverse transcription and quantitative real-time polymerase chain reaction were carried out using total RNA from blood samples of the proband and wild-type control subjects. A review of patients with OCNDS harboring CSNK2A1 pathogenic variants was conducted through a comprehensive search of the PubMed database.</p><p><strong>Results: </strong>We identified a novel CSNK2A1 frameshift variant p.Tyr323Leufs*16 in a Chinese family. The proband, a 31-year-old female, presented with abnormal eating habits, recurrent seizures, language impairment, and intellectual disability. Her mother exhibited postnatal hernias, splenomegaly, and a predisposition to infections, but showed no significant developmental impairments or intellectual disability. Genetic studies revealed the presence of this variant in CSNK2A1 in both the proband and her mother. Transcription analysis revealed this variant may lead to nonsense-mediated mRNA decay, suggesting haploinsufficiency as a potential disease mechanism. We reviewed 47 previously reported OCNDS cases and discovered that individuals carrying CSNK2A1 null variants may exhibit a diminished frequency of symptoms linked to language deficits, dysmorphic facial features, or intellectual disability, consequently presenting an overall milder phenotype when compared to those with missense variants.</p><p><strong>Conclusion: </strong>We report a novel frameshift variant, p.Tyr323Leufs*16, in an OCNDS family with a generally mild phenotype. This study may broaden the spectrum of clinical presentations associated with OCNDS and contribute novel insights into the genotype-phenotype correlation of this condition.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":null,"pages":null},"PeriodicalIF":1.5000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10915366/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2398","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Okur-Chung neurodevelopmental syndrome (OCNDS) is a rare autosomal dominant disorder caused by pathogenic variants in CSNK2A1. It is characterized by intellectual disability, developmental delay, and multisystemic abnormalities.
Methods: We performed the whole-exome sequencing for a patient in a Chinese family. The co-segregation study using the Sanger sequencing method was performed among family members. Reverse transcription and quantitative real-time polymerase chain reaction were carried out using total RNA from blood samples of the proband and wild-type control subjects. A review of patients with OCNDS harboring CSNK2A1 pathogenic variants was conducted through a comprehensive search of the PubMed database.
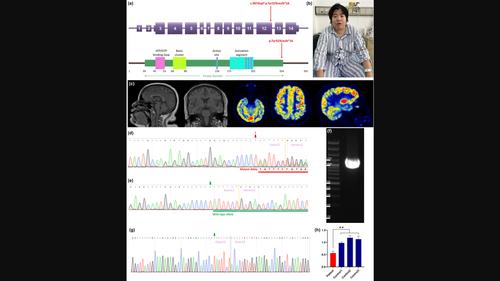
Results: We identified a novel CSNK2A1 frameshift variant p.Tyr323Leufs*16 in a Chinese family. The proband, a 31-year-old female, presented with abnormal eating habits, recurrent seizures, language impairment, and intellectual disability. Her mother exhibited postnatal hernias, splenomegaly, and a predisposition to infections, but showed no significant developmental impairments or intellectual disability. Genetic studies revealed the presence of this variant in CSNK2A1 in both the proband and her mother. Transcription analysis revealed this variant may lead to nonsense-mediated mRNA decay, suggesting haploinsufficiency as a potential disease mechanism. We reviewed 47 previously reported OCNDS cases and discovered that individuals carrying CSNK2A1 null variants may exhibit a diminished frequency of symptoms linked to language deficits, dysmorphic facial features, or intellectual disability, consequently presenting an overall milder phenotype when compared to those with missense variants.
Conclusion: We report a novel frameshift variant, p.Tyr323Leufs*16, in an OCNDS family with a generally mild phenotype. This study may broaden the spectrum of clinical presentations associated with OCNDS and contribute novel insights into the genotype-phenotype correlation of this condition.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


