Yvonne Mücke, Natalia Jablonka, Nicole Rimann, Hiu Man Grisch-Chan, Bernhard Hoffmann, Stefan Schillberg, Beat Thöny, Stefan Rasche
{"title":"A phenylalanine-free recombinant nutritional protein for the dietary management of phenylketonuria","authors":"Yvonne Mücke, Natalia Jablonka, Nicole Rimann, Hiu Man Grisch-Chan, Bernhard Hoffmann, Stefan Schillberg, Beat Thöny, Stefan Rasche","doi":"10.1002/jimd.12719","DOIUrl":null,"url":null,"abstract":"<p>Phenylketonuria (PKU) is a congenital metabolic disorder that causes the systemic elevation of phenylalanine (Phe), which is neurotoxic and teratogenic. PKU is currently incurable, and management involves lifelong adherence to an unpalatable protein-restricted diet based on Phe-free amino acid mixtures. Seeking a palatable dietary alternative, we identified a <i>Bacillus subtilis</i> protein (GSP16O) with a well-balanced but low-Phe amino acid profile. We optimized the sequence and expressed a modified Phe-free version (GSP105) in <i>Pseudomonas fluorescens</i>, achieving yields of 20 g/L. The purified GSP105 protein has a neutral taste and smell, is highly soluble, and remains stable up to 80°C. Homozygous <i>enu2</i> mice, a model of human PKU, were fed with diets containing either GSP105 or normal protein. The GSP105 diet led to normalization of blood Phe levels and brain monoamine neurotransmitter metabolites, and prevented maternal PKU. The GSP105 diet thus provides an alternative and efficacious dietary management strategy for PKU.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 4","pages":"651-663"},"PeriodicalIF":4.2000,"publicationDate":"2024-02-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12719","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12719","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
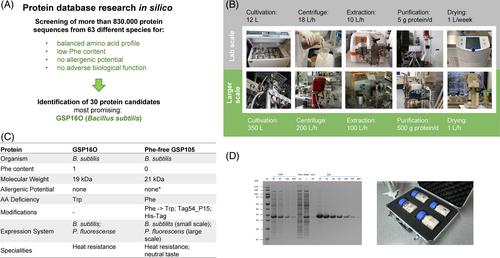
Phenylketonuria (PKU) is a congenital metabolic disorder that causes the systemic elevation of phenylalanine (Phe), which is neurotoxic and teratogenic. PKU is currently incurable, and management involves lifelong adherence to an unpalatable protein-restricted diet based on Phe-free amino acid mixtures. Seeking a palatable dietary alternative, we identified a Bacillus subtilis protein (GSP16O) with a well-balanced but low-Phe amino acid profile. We optimized the sequence and expressed a modified Phe-free version (GSP105) in Pseudomonas fluorescens, achieving yields of 20 g/L. The purified GSP105 protein has a neutral taste and smell, is highly soluble, and remains stable up to 80°C. Homozygous enu2 mice, a model of human PKU, were fed with diets containing either GSP105 or normal protein. The GSP105 diet led to normalization of blood Phe levels and brain monoamine neurotransmitter metabolites, and prevented maternal PKU. The GSP105 diet thus provides an alternative and efficacious dietary management strategy for PKU.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


