{"title":"The global low-energy structures of Al–Si eutectic and hypereutectic","authors":"Lin Zhang, Hongshan Chen","doi":"10.1007/s00214-024-03096-y","DOIUrl":null,"url":null,"abstract":"<p>The atomic-scale structures of Al–Si eutectics and hypereutectic are studied by using global structure searching method combined with ab initio density functional theories. The chemical components Al<sub>7</sub>Si, Al<sub>6</sub>Si and Al<sub>4</sub>Si, with silicon contents of 12.9, 14.7 and 20.6 wt%, are set for searching the lowest-energy structures. The global search results show that all of the low-energy structures demonstrate the fcc structure feature of pure aluminum crystal; while, the alloy structures distort slightly due to the addition of silicon. In the term of binding energy, the stabilities of the alloys enhance with increasing the silicon contents. The binding energies are approximated as the sum of the bond energies, and the least square fitting results give the Al–Al, Al–Si and Si–Si bond energies as − 0.624, − 0.731 and − 0.819 eV, respectively. Forming two Al–Si bonds sacrifices one Si–Si and one Al–Al bonds, and the Al–Si alloys have an energy gain of − 0.019 eV. It suggests that dispersion of silicon atoms in the alloys is favorable for the stability. The mechanical properties of the structures are calculated. While the bulk moduli of the alloys are very close, the shear and Young’s moduli are quite different for different structures with different distributions of silicon atoms.</p>","PeriodicalId":23045,"journal":{"name":"Theoretical Chemistry Accounts","volume":"33 1","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2024-02-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Chemistry Accounts","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1007/s00214-024-03096-y","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
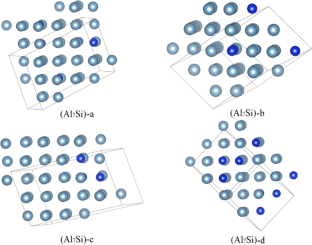
The atomic-scale structures of Al–Si eutectics and hypereutectic are studied by using global structure searching method combined with ab initio density functional theories. The chemical components Al7Si, Al6Si and Al4Si, with silicon contents of 12.9, 14.7 and 20.6 wt%, are set for searching the lowest-energy structures. The global search results show that all of the low-energy structures demonstrate the fcc structure feature of pure aluminum crystal; while, the alloy structures distort slightly due to the addition of silicon. In the term of binding energy, the stabilities of the alloys enhance with increasing the silicon contents. The binding energies are approximated as the sum of the bond energies, and the least square fitting results give the Al–Al, Al–Si and Si–Si bond energies as − 0.624, − 0.731 and − 0.819 eV, respectively. Forming two Al–Si bonds sacrifices one Si–Si and one Al–Al bonds, and the Al–Si alloys have an energy gain of − 0.019 eV. It suggests that dispersion of silicon atoms in the alloys is favorable for the stability. The mechanical properties of the structures are calculated. While the bulk moduli of the alloys are very close, the shear and Young’s moduli are quite different for different structures with different distributions of silicon atoms.
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


