Adsorption attributes of methyl naphthalene and naphthalene on P-Germanane sheets–a DFT outlook
Abstract
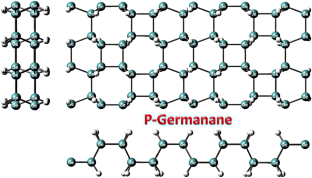
The adsorption attributes of methyl naphthalene and naphthalene molecules on P-Germanane sheets are explored based on the density functional theory method. The novel P-Germanane stability is established based on formation energy and phonon band spectrum. The stable P-Germanane exhibits a band gap of 3.944 eV. The calculated adsorption energy infers that methyl naphthalene and naphthalene are physisorbed on P-Germanane sheets. The charge transfer reveals that P-Germanane sheets behave as donors of electrons and target molecules behave as acceptors. The electronic attributes of P-Germanane sheets get altered owing to methyl naphthalene and naphthalene adsorption inferred from the band structure, PDOS spectrum, and electron density difference diagrams. We identified global minima position upon adsorption of 1-methylnaphthalene, and naphthalene on P-Germanane, which are named as bridge, octal, and tetra sites and adsorption attributes are explored in these sites. The adsorption energy and relative energy gap variation are noticed to be maximum for tetra site orientation of naphthalene on P-Germanane. The outcome exposes that P-Germanane sheets can be used as adsorption substrates for the detection of methyl naphthalene and naphthalene molecules.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


