Manli Zhao, Tingting Huang, Xueping Xiang, Yang Liu, Weizhong Gu, Lei Liu, Hongfeng Tang, Jinghong Xu, Jianhua Mao
{"title":"A 7-year-old boy presented with temporal lobe lesion","authors":"Manli Zhao, Tingting Huang, Xueping Xiang, Yang Liu, Weizhong Gu, Lei Liu, Hongfeng Tang, Jinghong Xu, Jianhua Mao","doi":"10.1111/bpa.13246","DOIUrl":null,"url":null,"abstract":"<p>A previously healthy 7-year-old boy presented with generalized tonic-clonic seizures for approximately 1 month. He was the first child of unrelated, healthy parents and had exhibited normal development. MR imaging demonstrated a large, right-sided temporal lobe mass-like lesion measuring 44 × 25 × 24 mm. The lesion exhibited hypointense on T1-weighted images and a distinct heterogenous high signal intensity on T2/FLAIR images with nodular contrast enhancement (Figure 1). He underwent surgical gross total resection of the tumor and postoperatively he was free of symptoms. Eight months post-surgery, neuroimaging gave no indication of tumor recurrence.</p><p>Histological examination (Box 1) unveiled a lesion with two distinct morphological components: a highly cellular area in the superficial cortex and an area characterized by sparse cells in the central region (Figure 2). The highly cellular area consisted of dense spindle-shaped tumor cells with round to oval or irregular nuclei and scant cytoplasm. These cells were set against an edematous, myxoid, or collagenous background, and were diffusely distributed throughout (Figure 2A). The hypocellular area housed scattered calcification foci and a fibrillary matrix with sparsely distributed, yet unremarkable, infiltrative tumor cells possessing oval nuclei (Figure 2B). Both components contained occasional interspersed degenerating neurons and large reactive astrocytes. No rhabdoid cells were present. Mitotic figures could be identified in some regions of high cellularity (up to 5 mitotic figures in 10 visual fields at magnification 400×), but not in the hypocellular areas. Necrosis was not found.</p><p>The tumor cells showed negative INI1 expression. Interestingly, the reactivity was preserved in the degenerating neurons and reactive astrocytes (Figure 2C, D). The tumor cells were diffusely positive for vimentin, partially positive for CD34, yet remained negative for GFAP, Olig2, EMA, AE1/AE3, αSMA, S-100 protein, and NeuN. The degenerating neurons showed positive NeuN expression, while the large reactive astrocytes demonstrated immunoreactivity against GFAP and vimentin. Ki-67 labeling indices were noted at around 15% and 0.5% in the hypercellular and hypocellular areas, respectively (Figure 2E, F).</p><p>Applying a DNA methylation-based classification, the tumor was classified as AT/RT-MYC (with calibrated scores of 0.96). A homozygous SMARCB1/INI1 deletion was indicated through copy number analysis using DNA methylation array data, and this was further validated by fluorescence in situ hybridization.</p><p>Low-grade diffusely infiltrative tumor (LGDIT), SMARCB1-deficient, with high-grade component.</p><p>Central nervous system LGDIT with SMARCB1/INI1-deficiency has been proposed as a new entity in recent literatures [<span>1, 2</span>]. Intriguingly, despite the loss of INI1 expression, it demonstrates distinct clinical and histopathological features, distinguishing it from atypical teratoid/rhabdoid tumors (AT/RT) [<span>1, 2</span>]. LGDIT, SMARCB1-deficient, predominantly occurs in supratentorial locations in older children and young adults, an age group where AT/RT is only sporadically encountered. Histopathologically, it generally shows low proliferative activity, a diffusely infiltrative growth pattern, and an absence of rhabdoid cells and polyphenotypic immunoreactivity. These characteristics can aid in differentiating it from AT/RT [<span>1, 2</span>]. Unlike AT/RT, LGDIT, SMARCB1-deficient, is typically reported as a clinically indolent tumor with a stable disease course for the majority of cases [<span>2</span>].</p><p>Most reported LGDIT, SMARCB1-deficient tumors exhibit low to moderate cellularity, with tumor cells displaying a low-grade morphology [<span>1, 2</span>]. In contrast, a minority of cases harbor a high-grade AT/RT component at the time of initial surgery, which is characterized by densely packed rhabdoid tumor cells with increased mitotic and proliferative activity [<span>1-3</span>]. The clinical implications of these high-grade components remain debated. While three LGDIT cases with the AT/RT component had a favorable outcome during follow-ups ranging from nine to 56 months [<span>2, 3</span>], another study reported two patients with primary and recurrent LGDIT containing high-grade AT/RT components; unfortunately, both patients succumbed to the disease within 3 and 41 months, respectively [<span>1</span>].</p><p>The DNA methylation profiles of all reported LGDIT, SMARCB1-deficient cases, including ours, consistently cluster close to the AT/RT-MYC group [<span>1-3</span>]. In addition, the two distinct morphological components revealed a close epigenetic resemblance [<span>3</span>]. Spatial transcriptome analyses highlighted that the transcriptional differences between the two components largely arise from variations in glioneuronal markers and extracellular matrix components [<span>3</span>].</p><p>A limited number of LGDIT, SMARCB1-deficient cases have been reported to progress to AT/RT after an observation period ranging from 1.5 to 7 years [<span>1</span>]. Given the relatively small sample size of the present series and the somewhat short follow-up time for most reported cases, it cannot be dismissed that all LGDIT, SMARCB1-deficient cases might eventually progress to AT/RT [<span>1-3</span>]. Consequently, careful follow-up examinations and consideration of the potential need for early therapeutic intervention are required.</p><p>Manli Zhao and Tingting Huang wrote the original draft. Xueping Xiang, Yang Liu, and Hongfeng Tang analyzed the data and co-wrote the manuscript. Weizhong Gu and Lei Liu performed the experiments. Jinghong Xu and Jianhua Mao revised the manuscript. All authors reviewed and approved the final manuscript.</p><p>This work was supported by grants from the Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology [Grant No. JBZX-202003].</p><p>The authors declare no conflicts of interest.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 3","pages":""},"PeriodicalIF":5.8000,"publicationDate":"2024-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13246","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13246","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
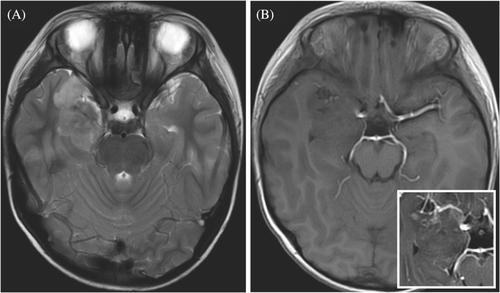
A previously healthy 7-year-old boy presented with generalized tonic-clonic seizures for approximately 1 month. He was the first child of unrelated, healthy parents and had exhibited normal development. MR imaging demonstrated a large, right-sided temporal lobe mass-like lesion measuring 44 × 25 × 24 mm. The lesion exhibited hypointense on T1-weighted images and a distinct heterogenous high signal intensity on T2/FLAIR images with nodular contrast enhancement (Figure 1). He underwent surgical gross total resection of the tumor and postoperatively he was free of symptoms. Eight months post-surgery, neuroimaging gave no indication of tumor recurrence.
Histological examination (Box 1) unveiled a lesion with two distinct morphological components: a highly cellular area in the superficial cortex and an area characterized by sparse cells in the central region (Figure 2). The highly cellular area consisted of dense spindle-shaped tumor cells with round to oval or irregular nuclei and scant cytoplasm. These cells were set against an edematous, myxoid, or collagenous background, and were diffusely distributed throughout (Figure 2A). The hypocellular area housed scattered calcification foci and a fibrillary matrix with sparsely distributed, yet unremarkable, infiltrative tumor cells possessing oval nuclei (Figure 2B). Both components contained occasional interspersed degenerating neurons and large reactive astrocytes. No rhabdoid cells were present. Mitotic figures could be identified in some regions of high cellularity (up to 5 mitotic figures in 10 visual fields at magnification 400×), but not in the hypocellular areas. Necrosis was not found.
The tumor cells showed negative INI1 expression. Interestingly, the reactivity was preserved in the degenerating neurons and reactive astrocytes (Figure 2C, D). The tumor cells were diffusely positive for vimentin, partially positive for CD34, yet remained negative for GFAP, Olig2, EMA, AE1/AE3, αSMA, S-100 protein, and NeuN. The degenerating neurons showed positive NeuN expression, while the large reactive astrocytes demonstrated immunoreactivity against GFAP and vimentin. Ki-67 labeling indices were noted at around 15% and 0.5% in the hypercellular and hypocellular areas, respectively (Figure 2E, F).
Applying a DNA methylation-based classification, the tumor was classified as AT/RT-MYC (with calibrated scores of 0.96). A homozygous SMARCB1/INI1 deletion was indicated through copy number analysis using DNA methylation array data, and this was further validated by fluorescence in situ hybridization.
Low-grade diffusely infiltrative tumor (LGDIT), SMARCB1-deficient, with high-grade component.
Central nervous system LGDIT with SMARCB1/INI1-deficiency has been proposed as a new entity in recent literatures [1, 2]. Intriguingly, despite the loss of INI1 expression, it demonstrates distinct clinical and histopathological features, distinguishing it from atypical teratoid/rhabdoid tumors (AT/RT) [1, 2]. LGDIT, SMARCB1-deficient, predominantly occurs in supratentorial locations in older children and young adults, an age group where AT/RT is only sporadically encountered. Histopathologically, it generally shows low proliferative activity, a diffusely infiltrative growth pattern, and an absence of rhabdoid cells and polyphenotypic immunoreactivity. These characteristics can aid in differentiating it from AT/RT [1, 2]. Unlike AT/RT, LGDIT, SMARCB1-deficient, is typically reported as a clinically indolent tumor with a stable disease course for the majority of cases [2].
Most reported LGDIT, SMARCB1-deficient tumors exhibit low to moderate cellularity, with tumor cells displaying a low-grade morphology [1, 2]. In contrast, a minority of cases harbor a high-grade AT/RT component at the time of initial surgery, which is characterized by densely packed rhabdoid tumor cells with increased mitotic and proliferative activity [1-3]. The clinical implications of these high-grade components remain debated. While three LGDIT cases with the AT/RT component had a favorable outcome during follow-ups ranging from nine to 56 months [2, 3], another study reported two patients with primary and recurrent LGDIT containing high-grade AT/RT components; unfortunately, both patients succumbed to the disease within 3 and 41 months, respectively [1].
The DNA methylation profiles of all reported LGDIT, SMARCB1-deficient cases, including ours, consistently cluster close to the AT/RT-MYC group [1-3]. In addition, the two distinct morphological components revealed a close epigenetic resemblance [3]. Spatial transcriptome analyses highlighted that the transcriptional differences between the two components largely arise from variations in glioneuronal markers and extracellular matrix components [3].
A limited number of LGDIT, SMARCB1-deficient cases have been reported to progress to AT/RT after an observation period ranging from 1.5 to 7 years [1]. Given the relatively small sample size of the present series and the somewhat short follow-up time for most reported cases, it cannot be dismissed that all LGDIT, SMARCB1-deficient cases might eventually progress to AT/RT [1-3]. Consequently, careful follow-up examinations and consideration of the potential need for early therapeutic intervention are required.
Manli Zhao and Tingting Huang wrote the original draft. Xueping Xiang, Yang Liu, and Hongfeng Tang analyzed the data and co-wrote the manuscript. Weizhong Gu and Lei Liu performed the experiments. Jinghong Xu and Jianhua Mao revised the manuscript. All authors reviewed and approved the final manuscript.
This work was supported by grants from the Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology [Grant No. JBZX-202003].
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


