{"title":"Chimeric Genes Causing 11β-Hydroxylase Deficiency: Implications in Clinical and Molecular Diagnosis.","authors":"Paola Concolino","doi":"10.1007/s40291-024-00697-y","DOIUrl":null,"url":null,"abstract":"<p><p>Deficiency of 11β-hydroxylase (11β-OHD) is the second most common cause of congenital adrenal hyperplasia (CAH), accounting for 0.2-8% of all cases. The disease is transmitted as an autosomal recessive trait and the underlying genetic causes of 11β-OHD are primarily small pathogenic variants affecting the CYP11B1 gene coding the 11β-hydroxylase enzyme. However, special events complicate the molecular diagnosis of 11β-OHD such as an unequal crossing over between the CYP11B2 (coding aldosterone synthase enzyme) and CYP11B1 genes. The resulting allele contains a hybrid gene, with a CYP11B2 5'-end and a CYP11B1 3'-end, where the CYP11B1 gene is under the control of the CYP11B2 promoter and thus not responding to the adrenocorticotropin (ACTH) but to angiotensin II and K<sup>+</sup>. This leads a reduction of cortisol production in 11β-OHD. In particular, CYP11B2/CYP11B1 chimeric genes can be distinguished into two groups depending on the breakpoint site: chimeras with breakpoint after the exon 5 of CYP11B2 preserve the aldosterone synthase activity, the others with breakpoint before exon 5 lose this function. In the last case, a more severe phenotype is expected. The aim of this review was to explore the setting of CYP11B2/CYP11B1 chimeras in 11β-OHD, performing a careful review of clinical literature cases.</p>","PeriodicalId":49797,"journal":{"name":"Molecular Diagnosis & Therapy","volume":" ","pages":"215-224"},"PeriodicalIF":4.1000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Diagnosis & Therapy","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40291-024-00697-y","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/7 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
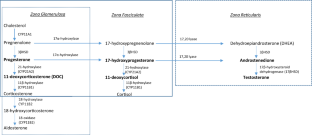
Deficiency of 11β-hydroxylase (11β-OHD) is the second most common cause of congenital adrenal hyperplasia (CAH), accounting for 0.2-8% of all cases. The disease is transmitted as an autosomal recessive trait and the underlying genetic causes of 11β-OHD are primarily small pathogenic variants affecting the CYP11B1 gene coding the 11β-hydroxylase enzyme. However, special events complicate the molecular diagnosis of 11β-OHD such as an unequal crossing over between the CYP11B2 (coding aldosterone synthase enzyme) and CYP11B1 genes. The resulting allele contains a hybrid gene, with a CYP11B2 5'-end and a CYP11B1 3'-end, where the CYP11B1 gene is under the control of the CYP11B2 promoter and thus not responding to the adrenocorticotropin (ACTH) but to angiotensin II and K+. This leads a reduction of cortisol production in 11β-OHD. In particular, CYP11B2/CYP11B1 chimeric genes can be distinguished into two groups depending on the breakpoint site: chimeras with breakpoint after the exon 5 of CYP11B2 preserve the aldosterone synthase activity, the others with breakpoint before exon 5 lose this function. In the last case, a more severe phenotype is expected. The aim of this review was to explore the setting of CYP11B2/CYP11B1 chimeras in 11β-OHD, performing a careful review of clinical literature cases.
期刊介绍:
Molecular Diagnosis & Therapy welcomes current opinion articles on emerging or contentious issues, comprehensive narrative reviews, systematic reviews (as outlined by the PRISMA statement), original research articles (including short communications) and letters to the editor. All manuscripts are subject to peer review by international experts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


