{"title":"Transient receptor potential vanilloid 1 inhibition reduces brain damage by suppressing neuronal apoptosis after intracerebral hemorrhage","authors":"Chien-Cheng Chen, Chia-Hua Ke, Chun-Hu Wu, Hung-Fu Lee, Yuan Chao, Min-Chien Tsai, Song-Kun Shyue, Szu-Fu Chen","doi":"10.1111/bpa.13244","DOIUrl":null,"url":null,"abstract":"<p>Intracerebral hemorrhage (ICH) induces a complex sequence of apoptotic cascades and inflammatory responses, leading to neurological impairment. Transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel with high calcium permeability, has been implicated in neuronal apoptosis and inflammatory responses. This study used a mouse ICH model and neuronal cultures to examine whether TRPV1 activation exacerbates brain damage and neurological deficits by promoting neuronal apoptosis and neuroinflammation. ICH was induced by injecting collagenase in both wild-type (WT) C57BL/6 mice and TRPV1<sup>−/−</sup> mice. Capsaicin (CAP; a TRPV1 agonist) or capsazepine (a TRPV1 antagonist) was administered by intracerebroventricular injection 30 min before ICH induction in WT mice. The effects of genetic deletion or pharmacological inhibition of TRPV1 using CAP or capsazepine on motor deficits, histological damage, apoptotic responses, blood–brain barrier (BBB) permeability, and neuroinflammatory reactions were explored. The antiapoptotic mechanisms and calcium influx induced by TRPV1 inactivation were investigated in cultured hemin-stimulated neurons. TRPV1 expression was upregulated in the hemorrhagic brain, and TRPV1 was expressed in neurons, microglia, and astrocytes after ICH. Genetic deletion of TRPV1 significantly attenuated motor deficits and brain atrophy for up to 28 days. Deletion of TRPV1 also reduced brain damage, neurodegeneration, microglial activation, cytokine expression, and cell apoptosis at 1 day post-ICH. Similarly, the administration of CAP ameliorated brain damage, neurodegeneration, brain edema, BBB permeability, and cytokine expression at 1 day post-ICH. In primary neuronal cultures, pharmacological inactivation of TRPV1 by CAP attenuated neuronal vulnerability to hemin-induced injury, suppressed apoptosis, and preserved mitochondrial integrity in vitro. Mechanistically, CAP reduced hemin-stimulated calcium influx and prevented the phosphorylation of CaMKII in cultured neurons, which was associated with reduced activation of P38 and c-Jun NH<sub>2</sub>-terminal kinase mitogen-activated protein kinase signaling. Our results suggest that TRPV1 inhibition may be a potential therapy for ICH by suppressing mitochondria-related neuronal apoptosis.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 5","pages":""},"PeriodicalIF":5.8000,"publicationDate":"2024-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13244","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13244","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
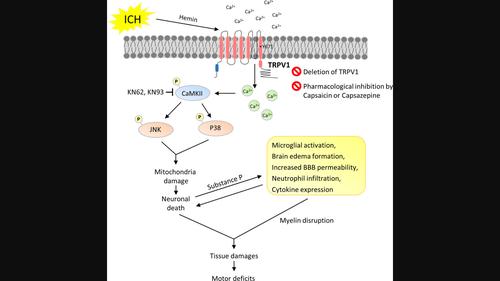
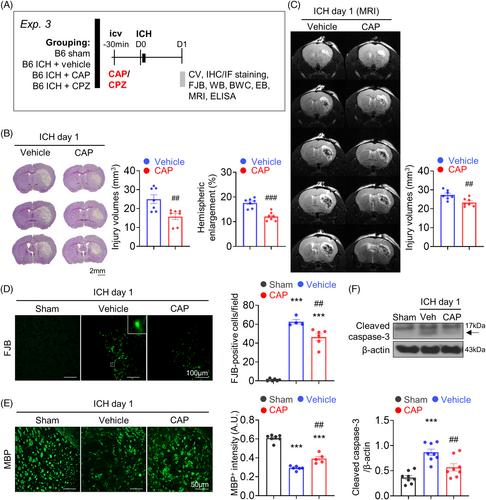
Intracerebral hemorrhage (ICH) induces a complex sequence of apoptotic cascades and inflammatory responses, leading to neurological impairment. Transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel with high calcium permeability, has been implicated in neuronal apoptosis and inflammatory responses. This study used a mouse ICH model and neuronal cultures to examine whether TRPV1 activation exacerbates brain damage and neurological deficits by promoting neuronal apoptosis and neuroinflammation. ICH was induced by injecting collagenase in both wild-type (WT) C57BL/6 mice and TRPV1−/− mice. Capsaicin (CAP; a TRPV1 agonist) or capsazepine (a TRPV1 antagonist) was administered by intracerebroventricular injection 30 min before ICH induction in WT mice. The effects of genetic deletion or pharmacological inhibition of TRPV1 using CAP or capsazepine on motor deficits, histological damage, apoptotic responses, blood–brain barrier (BBB) permeability, and neuroinflammatory reactions were explored. The antiapoptotic mechanisms and calcium influx induced by TRPV1 inactivation were investigated in cultured hemin-stimulated neurons. TRPV1 expression was upregulated in the hemorrhagic brain, and TRPV1 was expressed in neurons, microglia, and astrocytes after ICH. Genetic deletion of TRPV1 significantly attenuated motor deficits and brain atrophy for up to 28 days. Deletion of TRPV1 also reduced brain damage, neurodegeneration, microglial activation, cytokine expression, and cell apoptosis at 1 day post-ICH. Similarly, the administration of CAP ameliorated brain damage, neurodegeneration, brain edema, BBB permeability, and cytokine expression at 1 day post-ICH. In primary neuronal cultures, pharmacological inactivation of TRPV1 by CAP attenuated neuronal vulnerability to hemin-induced injury, suppressed apoptosis, and preserved mitochondrial integrity in vitro. Mechanistically, CAP reduced hemin-stimulated calcium influx and prevented the phosphorylation of CaMKII in cultured neurons, which was associated with reduced activation of P38 and c-Jun NH2-terminal kinase mitogen-activated protein kinase signaling. Our results suggest that TRPV1 inhibition may be a potential therapy for ICH by suppressing mitochondria-related neuronal apoptosis.
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


