Rescue of secretion of rare-disease-associated misfolded mutant glycoproteins in UGGT1 knock-out mammalian cells
IF 2.5
3区 生物学
Q3 CELL BIOLOGY
引用次数: 0
Abstract
Endoplasmic reticulum (ER) retention of misfolded glycoproteins is mediated by the ER-localized eukaryotic glycoprotein secretion checkpoint, UDP-glucose glycoprotein glucosyl-transferase (UGGT). The enzyme recognizes a misfolded glycoprotein and flags it for ER retention by re-glucosylating one of its N-linked glycans. In the background of a congenital mutation in a secreted glycoprotein gene, UGGT-mediated ER retention can cause rare disease, even if the mutant glycoprotein retains activity (“responsive mutant”). Using confocal laser scanning microscopy, we investigated here the subcellular localization of the human Trop-2-Q118E, E227K and L186P mutants, which cause gelatinous drop-like corneal dystrophy (GDLD). Compared with the wild-type Trop-2, which is correctly localized at the plasma membrane, these Trop-2 mutants are retained in the ER. We studied fluorescent chimeras of the Trop-2 Q118E, E227K and L186P mutants in mammalian cells harboring CRISPR/Cas9-mediated inhibition of the UGGT1 and/or UGGT2 genes. The membrane localization of the Trop-2 Q118E, E227K and L186P mutants was successfully rescued in UGGT1−/−cells. UGGT1 also efficiently reglucosylated Trop-2-Q118E-EYFP in cellula. The study supports the hypothesis that UGGT1 modulation would constitute a novel therapeutic strategy for the treatment of pathological conditions associated to misfolded membrane glycoproteins (whenever the mutation impairs but does not abrogate function), and it encourages the testing of modulators of ER glycoprotein folding quality control as broad-spectrum rescue-of-secretion drugs in rare diseases caused by responsive secreted glycoprotein mutants.
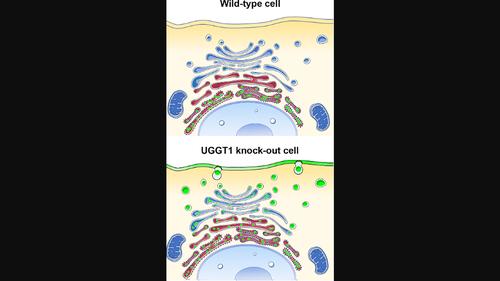
在 UGGT1 基因被敲除的哺乳动物细胞中挽救与罕见疾病相关的错误折叠突变糖蛋白的分泌
内质网(ER)保留折叠错误的糖蛋白是由位于ER的真核糖蛋白分泌检查点--UDP-葡萄糖糖蛋白葡萄糖基转移酶(UGGT)--介导的。这种酶能识别折叠错误的糖蛋白,并通过对其中一个 N-连接的糖基进行再糖基化,将其标记为保留在 ER 中。在分泌型糖蛋白基因先天突变的背景下,UGGT 介导的 ER 保留可导致罕见疾病,即使突变型糖蛋白仍具有活性("反应突变体")。利用激光共聚焦扫描显微镜,我们研究了人Trop-2-Q118E、E227K和L186P突变体的亚细胞定位,这些突变体会导致胶样滴状角膜营养不良症(GDLD)。与野生型 Trop-2 正确定位于质膜相比,这些 Trop-2 突变体保留在 ER 中。我们在通过 CRISPR/Cas9 介导抑制 UGGT1 和/或 UGGT2 基因的哺乳动物细胞中研究了 Trop-2 Q118E、E227K 和 L186P 突变体的荧光嵌合体。在 UGGT1-/ 细胞中,Trop-2 Q118E、E227K 和 L186P 突变体的膜定位被成功挽救。UGGT1 还能在细胞中有效地对 Trop-2-Q118E-EYFP 进行再葡糖基化。这项研究支持这样的假设,即调节 UGGT1 将成为一种新的治疗策略,用于治疗与折叠错误的膜糖蛋白相关的病理状况(只要突变会损害但不会削弱功能),它还鼓励将 ER 糖蛋白折叠质量控制调节剂作为广谱的分泌拯救药物,用于测试由反应性分泌糖蛋白突变体引起的罕见疾病。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Traffic
生物-细胞生物学
CiteScore
8.10
自引率
2.20%
发文量
50
审稿时长
2 months
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


