Julien Baruteau, Nandaki Keshavan, Charles P. Venditti
{"title":"Mission possible: Gene therapy for inherited metabolic diseases","authors":"Julien Baruteau, Nandaki Keshavan, Charles P. Venditti","doi":"10.1002/jimd.12708","DOIUrl":null,"url":null,"abstract":"<p>Since the description of ‘inborn errors of metabolism’ as a novel field of medicine by Archibald Garrod in 1908,<span><sup>1</sup></span> various breakthroughs in management and therapeutic milestones have been achieved: specific diets, newborn screening and enzyme replacement therapy to name a few (Figure 1). Genomic assays, including exome, genome and RNA sequencing, have led to the identification of a rapidly growing number of new inborn errors of metabolism and many new patients in recent years.</p><p>Gene therapy centred around gene addition and editing therapy has emerged in parallel with the technological progress in engineering nucleic acids, nucleases and viruses. Seminal early milestones have raised a huge hope for inherited metabolic diseases (IMDs) with little or no therapeutic benefit under standard of care.<span><sup>2, 3</sup></span> Complex biotechnologies such as gene addition mediated by adeno-associated viral vectors (AAV) and integrating vectors that rely upon lentiviral and CRISPR-Cas9 mediated gene-editing platforms are now the basis of approved drug products for monogenic diseases.<span><sup>4-6</sup></span> Application of gene therapy has been studied in many rare IMDs. Proof-of-concept data using varied technologies, nucleic acids, and delivery platforms to achieve gene replacement, integration and editing, especially in the liver and the central nervous system, have served to enable a wide range of exciting new therapies for genetic and metabolic disorders. Whilst first-in-man clinical trials expand, the challenges for this rapidly evolving field include the development of safer and more efficient vectors, more accessible technologies, and the development of new regulatory paradigms to expedite approvals. Today, a one-size-fits-all strategy remains elusive for most disorders given that even within a rare IEM patient population, phenotypic heterogeneity, variable disease progression and uncertainties surrounding the natural history can further complicate the risk–benefit balance for clinical trials.</p><p>This themed issue of <i>Journal of Inherited Metabolic Disease</i> reviews state-of-the-art of gene therapy technologies applied to various inborn metabolic diseases. It provides updates on clinical successes, limitations and future directions whilst considering specificities for liver and fetal applications. The special issue starts with two reviews concerning liver-directed gene therapy. Baruteau et al. present an overview of the progress, challenges and perspectives for the main liver IMDs from a clinical perspective.<span><sup>7</sup></span> Chuecos and Lagor introduce AAV, which represent currently the leading liver-targeting gene therapy technology, with a particular focus on AAV physiology, AAV transduction including sex differences and an updated review of AAV clinical trials for liver IMDs and their contribution to the field of gene therapy.<span><sup>8</sup></span> Pontoizeau et al. provide an additional proof of concept of neonatal gene therapy for maple syrup urine disease.<span><sup>9</sup></span> Duff et al. review the specificities, development and perspectives of gene therapy for urea cycle defects.<span><sup>10</sup></span> Chandler and Venditti summarise the preclinical studies and clinical trials of genetic therapies developed for methylmalonic and propionic acidaemias and lessons learned over the years.<span><sup>11</sup></span> Martinez et al. provide an update on proof of concept in animal models and clinical trials of gene therapy for phenylketonuria, including recent innovative preclinical approaches of introducing an acquired competitive advantage of genetically modified hepatocytes.<span><sup>12</sup></span> Koeberl et al. review gene therapy advances for glycogen storage diseases (GSDs) from preclinical to clinical studies, with a particular focus on GSDI, II and III.<span><sup>13</sup></span> Sellier et al. report a muscle-specific, liver-detargeted AAV gene therapy to treat a neonatal and adult Pompe mouse model.<span><sup>14</sup></span> Rossi and Brunetti-Pierri review the development of ex vivo and in vivo gene therapy approaches for mucopolysaccharidosis (MPS) with detailed sections for MPSI, MPSII, MPSIII, MPSIV and MPSVI.<span><sup>15</sup></span> Keshavan et al. provide a perspective on the development of gene therapy for primary mitochondrial diseases.<span><sup>16</sup></span> Ng et al. show the epic journey to develop gene therapy approaches for inherited neurotransmitter defects culminating with the recent approval of eladocagene exuparvovec (Upstaza™) for aromatic L-amino acid decarboxylase (AADC) deficiency in late 2022.<span><sup>17</sup></span> To conclude this special issue, Waddington et al. present a magistral review of fetal gene therapy and its complex implications from preclinical evidence to translation, safety and ethics for both the fetus and the mother.<span><sup>18</sup></span></p><p>In conclusion, numerous proof-of-concept studies have investigated different gene therapy technologies in preclinical models of IMDs. The wider translation of these promising therapeutic strategies to patients, and overcoming their inherent limitations, remains the bottleneck. Supported by improved knowledge gathered from numerous ongoing clinical trials for inborn errors of metabolism, the translation of innovative gene therapy for rare diseases, which remains a long and arduous journey, is gradually reaching patients, and is uplifted by the hope of a brighter outcome and a much-improved quality of life.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 1","pages":"5-6"},"PeriodicalIF":4.2000,"publicationDate":"2024-01-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12708","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12708","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
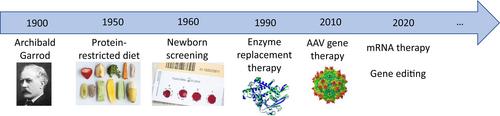
Since the description of ‘inborn errors of metabolism’ as a novel field of medicine by Archibald Garrod in 1908,1 various breakthroughs in management and therapeutic milestones have been achieved: specific diets, newborn screening and enzyme replacement therapy to name a few (Figure 1). Genomic assays, including exome, genome and RNA sequencing, have led to the identification of a rapidly growing number of new inborn errors of metabolism and many new patients in recent years.
Gene therapy centred around gene addition and editing therapy has emerged in parallel with the technological progress in engineering nucleic acids, nucleases and viruses. Seminal early milestones have raised a huge hope for inherited metabolic diseases (IMDs) with little or no therapeutic benefit under standard of care.2, 3 Complex biotechnologies such as gene addition mediated by adeno-associated viral vectors (AAV) and integrating vectors that rely upon lentiviral and CRISPR-Cas9 mediated gene-editing platforms are now the basis of approved drug products for monogenic diseases.4-6 Application of gene therapy has been studied in many rare IMDs. Proof-of-concept data using varied technologies, nucleic acids, and delivery platforms to achieve gene replacement, integration and editing, especially in the liver and the central nervous system, have served to enable a wide range of exciting new therapies for genetic and metabolic disorders. Whilst first-in-man clinical trials expand, the challenges for this rapidly evolving field include the development of safer and more efficient vectors, more accessible technologies, and the development of new regulatory paradigms to expedite approvals. Today, a one-size-fits-all strategy remains elusive for most disorders given that even within a rare IEM patient population, phenotypic heterogeneity, variable disease progression and uncertainties surrounding the natural history can further complicate the risk–benefit balance for clinical trials.
This themed issue of Journal of Inherited Metabolic Disease reviews state-of-the-art of gene therapy technologies applied to various inborn metabolic diseases. It provides updates on clinical successes, limitations and future directions whilst considering specificities for liver and fetal applications. The special issue starts with two reviews concerning liver-directed gene therapy. Baruteau et al. present an overview of the progress, challenges and perspectives for the main liver IMDs from a clinical perspective.7 Chuecos and Lagor introduce AAV, which represent currently the leading liver-targeting gene therapy technology, with a particular focus on AAV physiology, AAV transduction including sex differences and an updated review of AAV clinical trials for liver IMDs and their contribution to the field of gene therapy.8 Pontoizeau et al. provide an additional proof of concept of neonatal gene therapy for maple syrup urine disease.9 Duff et al. review the specificities, development and perspectives of gene therapy for urea cycle defects.10 Chandler and Venditti summarise the preclinical studies and clinical trials of genetic therapies developed for methylmalonic and propionic acidaemias and lessons learned over the years.11 Martinez et al. provide an update on proof of concept in animal models and clinical trials of gene therapy for phenylketonuria, including recent innovative preclinical approaches of introducing an acquired competitive advantage of genetically modified hepatocytes.12 Koeberl et al. review gene therapy advances for glycogen storage diseases (GSDs) from preclinical to clinical studies, with a particular focus on GSDI, II and III.13 Sellier et al. report a muscle-specific, liver-detargeted AAV gene therapy to treat a neonatal and adult Pompe mouse model.14 Rossi and Brunetti-Pierri review the development of ex vivo and in vivo gene therapy approaches for mucopolysaccharidosis (MPS) with detailed sections for MPSI, MPSII, MPSIII, MPSIV and MPSVI.15 Keshavan et al. provide a perspective on the development of gene therapy for primary mitochondrial diseases.16 Ng et al. show the epic journey to develop gene therapy approaches for inherited neurotransmitter defects culminating with the recent approval of eladocagene exuparvovec (Upstaza™) for aromatic L-amino acid decarboxylase (AADC) deficiency in late 2022.17 To conclude this special issue, Waddington et al. present a magistral review of fetal gene therapy and its complex implications from preclinical evidence to translation, safety and ethics for both the fetus and the mother.18
In conclusion, numerous proof-of-concept studies have investigated different gene therapy technologies in preclinical models of IMDs. The wider translation of these promising therapeutic strategies to patients, and overcoming their inherent limitations, remains the bottleneck. Supported by improved knowledge gathered from numerous ongoing clinical trials for inborn errors of metabolism, the translation of innovative gene therapy for rare diseases, which remains a long and arduous journey, is gradually reaching patients, and is uplifted by the hope of a brighter outcome and a much-improved quality of life.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


