Yujia Huang, Ming Tang, Zhiyi Hu, Bailian Cai, Guofang Chen, Lijun Jiang, Yan Xia, Pujun Guan, Xiaoqi Li, Zhiyong Mao, Xiaoping Wan, Wen Lu
{"title":"SMYD3 promotes endometrial cancer through epigenetic regulation of LIG4/XRCC4/XLF complex in non-homologous end joining repair","authors":"Yujia Huang, Ming Tang, Zhiyi Hu, Bailian Cai, Guofang Chen, Lijun Jiang, Yan Xia, Pujun Guan, Xiaoqi Li, Zhiyong Mao, Xiaoping Wan, Wen Lu","doi":"10.1038/s41389-023-00503-0","DOIUrl":null,"url":null,"abstract":"<p>Endometrial cancer (EC) stands as one of the most prevalent malignancies affecting the female genital tract, witnessing a rapid surge in incidence globally. Despite the well-established association of histone methyltransferase SMYD3 with the development and progression of various cancers, its specific oncogenic role in endometrial cancer remains unexplored. In the present study, we report that the expression level of SMYD3 is significantly upregulated in EC samples and associated with EC progression. Through meticulous in vivo and in vitro experiments, we reveal that depletion of SMYD3 curtails cell proliferation, migration, and invasion capabilities, leading to compromised non-homologous end joining repair (NHEJ) and heightened sensitivity of EC cells to radiation. Furthermore, our pathway enrichment analysis underscores the pivotal involvement of the DNA damage repair pathway in regulating EC progression. Mechanistically, in response to DNA damage, SMYD3 is recruited to these sites in a PARP1-dependent manner, specifically methylating LIG4. This methylation sets off a sequential assembly of the LIG4/XRCC4/XLF complex, actively participating in the NHEJ pathway and thereby fostering EC progression. Notably, our findings highlight the promise of SMYD3 as a crucial player in NHEJ repair and its direct correlation with EC progression. Intriguingly, pharmacological intervention targeting SMYD3 with its specific inhibitor, BCI-121, emerges as a potent strategy, markedly suppressing the tumorigenicity of EC cells and significantly enhancing the efficacy of radiotherapy. Collectively, our comprehensive data position SMYD3 as a central factor in NHEJ repair and underscore its potential as a promising pharmacological target for endometrial cancer therapy, validated through both in vitro and in vivo systems.</p>","PeriodicalId":19489,"journal":{"name":"Oncogenesis","volume":"34 1","pages":""},"PeriodicalIF":6.4000,"publicationDate":"2024-01-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Oncogenesis","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41389-023-00503-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
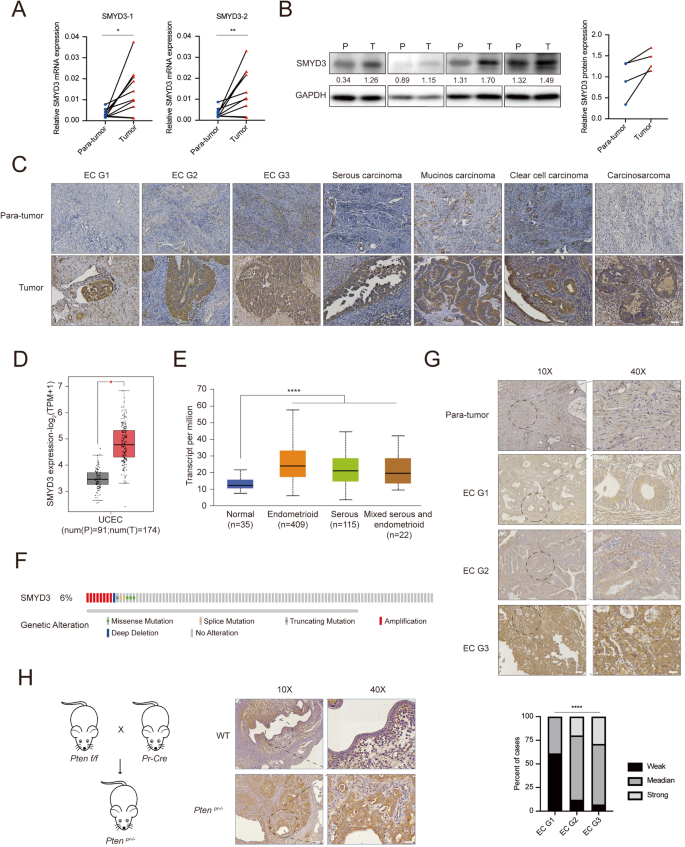
Endometrial cancer (EC) stands as one of the most prevalent malignancies affecting the female genital tract, witnessing a rapid surge in incidence globally. Despite the well-established association of histone methyltransferase SMYD3 with the development and progression of various cancers, its specific oncogenic role in endometrial cancer remains unexplored. In the present study, we report that the expression level of SMYD3 is significantly upregulated in EC samples and associated with EC progression. Through meticulous in vivo and in vitro experiments, we reveal that depletion of SMYD3 curtails cell proliferation, migration, and invasion capabilities, leading to compromised non-homologous end joining repair (NHEJ) and heightened sensitivity of EC cells to radiation. Furthermore, our pathway enrichment analysis underscores the pivotal involvement of the DNA damage repair pathway in regulating EC progression. Mechanistically, in response to DNA damage, SMYD3 is recruited to these sites in a PARP1-dependent manner, specifically methylating LIG4. This methylation sets off a sequential assembly of the LIG4/XRCC4/XLF complex, actively participating in the NHEJ pathway and thereby fostering EC progression. Notably, our findings highlight the promise of SMYD3 as a crucial player in NHEJ repair and its direct correlation with EC progression. Intriguingly, pharmacological intervention targeting SMYD3 with its specific inhibitor, BCI-121, emerges as a potent strategy, markedly suppressing the tumorigenicity of EC cells and significantly enhancing the efficacy of radiotherapy. Collectively, our comprehensive data position SMYD3 as a central factor in NHEJ repair and underscore its potential as a promising pharmacological target for endometrial cancer therapy, validated through both in vitro and in vivo systems.
期刊介绍:
Oncogenesis is a peer-reviewed open access online journal that publishes full-length papers, reviews, and short communications exploring the molecular basis of cancer and related phenomena. It seeks to promote diverse and integrated areas of molecular biology, cell biology, oncology, and genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


