{"title":"AlphaMissense, a groundbreaking advancement in artificial intelligence for predicting the effects of missense variants","authors":"Ming Yi, Yunqiang Liu, Zhiguang Su","doi":"10.1002/mef2.70","DOIUrl":null,"url":null,"abstract":"<p>In a recent study published in <i>Science</i>,<span><sup>1</sup></span> Cheng and colleagues developed a highly accurate protein structuring model named AlphaMissense, which can predict and characterize the pathogenicity of all possible missense variants in the human genome at a single amino acid substitution level. As a community resource, AlphaMissense is absolutely helping us to gain better insights into the functional consequences of genetic variation.</p><p>Despite the identification of over 4 million missense variants in the human genome, only approximately 2% are definitively annotated as pathogenic or benign, and the significance of the large proportion of missense variants is unknown. As such, there has been a push to search for highly effective methods to accurately predict the variants' clinical implications.</p><p>Presently, four primary methodologies have been used to predict the pathogenicity of genetic variations. The first class of methods is known as “database-driven approaches,” which rely extensively on meticulously curated databases. Such strategies suffer from data leakage caused by unintended information transfer between the training and test halves, posing a significant challenge to reliability and accuracy.<span><sup>1, 2</sup></span> The second class of methods is referred to as “weak-labeling approaches,” which circumvent circularity concerns by eliminating human annotations. However, such models often encounter false labels in the training data, necessitating the use of more reliable labels for accurate evaluation. A third class of approaches focuses on the recognition of naturally evolved amino acid sequence distributions and hidden structures of proteins, providing insights into the evolutionary patterns and functional characteristics of proteins.<span><sup>1</sup></span> Such models, however, do not possess the advanced understanding of protein structure achieved by AlphaFold (AF).<span><sup>3</sup></span> A fourth approach utilizes protein structure information to improve the assessment of genetic constraints. However, this approach encounters a new challenge in the accuracy of predicting variant pathogenicity based solely on structural features. The limited performance of the structure-based approach in predicting pathogenicity in ClinVar variations suggests that additional factors, such as functional annotations as well as population frequencies and clinical evidence, play a crucial role in determining the pathogenicity of a genetic variant.<span><sup>1</sup></span></p><p>AlphaMissense, constructed upon the protein structure prediction model of AF, is a machine-learning model that utilizes advancements in unsupervised protein language modeling (Figure 1). AF represents a groundbreaking method that enables the prediction of a protein's three-dimensional structure solely from its amino acid sequence.<span><sup>3</sup></span> By incorporating the structural insights provided by AF, researchers have achieved notable advancements in accurately assessing the potential pathogenic impact of genetic variations.<span><sup>4</sup></span></p><p>AlphaMissense incorporates structural context from AF-derived systems and fine-tunes using weak labels obtained from population frequency data. Remarkably, this model achieves state-of-the-art predictions in clinical annotation, identification of de novo disease variants, and experimental benchmarks, even without specific training on datasets tailored to these tasks. One notable feature of AlphaMissense lies in its capacity to provide predictions for a wide range of genetic variations, particularly those that have not been supported by experimental data. AlphaMissense fills this gap by providing predictions for these variants, enabling researchers to gain insights into their potential functional consequences.</p><p>In addition to the capacity to predict pathogenicity, AlphaMissense has shown potential in predicting the essentiality of a gene for cell survival or fitness. The databases produced by AlphaMissense are accessible to the scientific community, providing valuable resources that include forecasts unique to 60,000 alternative transcripts.<span><sup>1</sup></span> The availability of accurate predictions for single-amino acid alterations through AlphaMissence empowers researchers to prioritize variants for further experimental investigation, reducing the need for extensive and costly experimental characterization of each variant. This accelerates the research process and enables a more efficient allocation of resources.</p><p>While AlphaMissense exhibits efficacy in predicting pathogenicity via scalar values, certain limitations warrant consideration. First, AlphaMissense utilizes wild-type structural predictions but does not directly provide detailed structural change information for altered sequences. Additional analyses and techniques are needed to investigate structural implications. Integrating multimodal and experimental data remains imperative to obtain a comprehensive understanding of structural ramifications and influences on protein stability and function. Second, AlphaMissense does not explicitly predict missense variants' impacts on biophysical properties, like, stability and binding affinity. Third, AlphaMissense does not incorporate training to account for potential interactions with other proteins during pathogenicity prediction. It is also limited to single amino acid substitutions, not encompassing more complex variations. Performance may be limited for de novo proteins lacking evolutionary information, as the model relies heavily on multiple sequence alignments for structure prediction and evolutionary conservation estimation. Additionally, it is also important to acknowledge calibration score deviations from expected probabilities observed in ClinVar, suggesting that predicted probabilities may not always perfectly align with clinical pathogenicity observations. These limitations underscore the need for further modeling advances to address more complex variations and improve pathogenicity prediction for de novo proteins. Ultimately, it is important to note that the concept of mutation pathogenicity is exceedingly complex with numerous contributing factors. Computational prediction models often oversimplify this complexity, omitting important considerations, such as inheritance pattern, allelic state, and incomplete penetrance.<span><sup>5</sup></span> In particular, the phenomenon of incomplete penetrance underscores that not all carriers of a purportedly “pathogenic” allele will necessarily manifest clinical disease. Rather, one's genetic background and environmental exposures can modulate the phenotypic effects of mutations. Fully accounting for such nuances remains a notable challenge for in silico approaches. A more comprehensive integration of diverse data, including family history, genetic context, and environmental interactions, may help advance computational pathogenicity predictions by more closely approximating the intricate interplay of biological and environmental determinants that collectively influence disease risk and expression in the real world.</p><p>In summary, as a significantly noteworthy advancement in the field of protein variant analysis, AlphaMissense predicts the possible impact of every amino acid substitution in the human proteome and classifies 89% of missense variants as either likely benign or likely pathogenic. In addition, AlphaMissense interprets the effects of every possible missense mutation in the human genome on protein structure and function based on the insights obtained from protein structure analysis. Overall, by incorporating the pathogenic classification of missense mutations of unknown significance into protein structure-predicting models, AlphaMissense holds promise to identify where disease-causing mutations are likely to occur in a protein.</p><p>Ming Yi and Yunqiang Liu drafted the manuscript, Zhiguang Su revised the manuscript. All authors have read and approved the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>Not applicable.</p>","PeriodicalId":74135,"journal":{"name":"MedComm - Future medicine","volume":"3 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-01-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.70","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm - Future medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mef2.70","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
In a recent study published in Science,1 Cheng and colleagues developed a highly accurate protein structuring model named AlphaMissense, which can predict and characterize the pathogenicity of all possible missense variants in the human genome at a single amino acid substitution level. As a community resource, AlphaMissense is absolutely helping us to gain better insights into the functional consequences of genetic variation.
Despite the identification of over 4 million missense variants in the human genome, only approximately 2% are definitively annotated as pathogenic or benign, and the significance of the large proportion of missense variants is unknown. As such, there has been a push to search for highly effective methods to accurately predict the variants' clinical implications.
Presently, four primary methodologies have been used to predict the pathogenicity of genetic variations. The first class of methods is known as “database-driven approaches,” which rely extensively on meticulously curated databases. Such strategies suffer from data leakage caused by unintended information transfer between the training and test halves, posing a significant challenge to reliability and accuracy.1, 2 The second class of methods is referred to as “weak-labeling approaches,” which circumvent circularity concerns by eliminating human annotations. However, such models often encounter false labels in the training data, necessitating the use of more reliable labels for accurate evaluation. A third class of approaches focuses on the recognition of naturally evolved amino acid sequence distributions and hidden structures of proteins, providing insights into the evolutionary patterns and functional characteristics of proteins.1 Such models, however, do not possess the advanced understanding of protein structure achieved by AlphaFold (AF).3 A fourth approach utilizes protein structure information to improve the assessment of genetic constraints. However, this approach encounters a new challenge in the accuracy of predicting variant pathogenicity based solely on structural features. The limited performance of the structure-based approach in predicting pathogenicity in ClinVar variations suggests that additional factors, such as functional annotations as well as population frequencies and clinical evidence, play a crucial role in determining the pathogenicity of a genetic variant.1
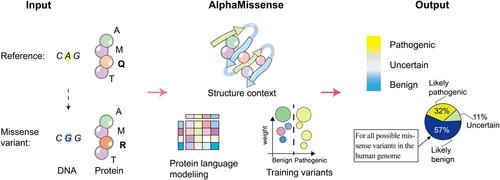
AlphaMissense, constructed upon the protein structure prediction model of AF, is a machine-learning model that utilizes advancements in unsupervised protein language modeling (Figure 1). AF represents a groundbreaking method that enables the prediction of a protein's three-dimensional structure solely from its amino acid sequence.3 By incorporating the structural insights provided by AF, researchers have achieved notable advancements in accurately assessing the potential pathogenic impact of genetic variations.4
AlphaMissense incorporates structural context from AF-derived systems and fine-tunes using weak labels obtained from population frequency data. Remarkably, this model achieves state-of-the-art predictions in clinical annotation, identification of de novo disease variants, and experimental benchmarks, even without specific training on datasets tailored to these tasks. One notable feature of AlphaMissense lies in its capacity to provide predictions for a wide range of genetic variations, particularly those that have not been supported by experimental data. AlphaMissense fills this gap by providing predictions for these variants, enabling researchers to gain insights into their potential functional consequences.
In addition to the capacity to predict pathogenicity, AlphaMissense has shown potential in predicting the essentiality of a gene for cell survival or fitness. The databases produced by AlphaMissense are accessible to the scientific community, providing valuable resources that include forecasts unique to 60,000 alternative transcripts.1 The availability of accurate predictions for single-amino acid alterations through AlphaMissence empowers researchers to prioritize variants for further experimental investigation, reducing the need for extensive and costly experimental characterization of each variant. This accelerates the research process and enables a more efficient allocation of resources.
While AlphaMissense exhibits efficacy in predicting pathogenicity via scalar values, certain limitations warrant consideration. First, AlphaMissense utilizes wild-type structural predictions but does not directly provide detailed structural change information for altered sequences. Additional analyses and techniques are needed to investigate structural implications. Integrating multimodal and experimental data remains imperative to obtain a comprehensive understanding of structural ramifications and influences on protein stability and function. Second, AlphaMissense does not explicitly predict missense variants' impacts on biophysical properties, like, stability and binding affinity. Third, AlphaMissense does not incorporate training to account for potential interactions with other proteins during pathogenicity prediction. It is also limited to single amino acid substitutions, not encompassing more complex variations. Performance may be limited for de novo proteins lacking evolutionary information, as the model relies heavily on multiple sequence alignments for structure prediction and evolutionary conservation estimation. Additionally, it is also important to acknowledge calibration score deviations from expected probabilities observed in ClinVar, suggesting that predicted probabilities may not always perfectly align with clinical pathogenicity observations. These limitations underscore the need for further modeling advances to address more complex variations and improve pathogenicity prediction for de novo proteins. Ultimately, it is important to note that the concept of mutation pathogenicity is exceedingly complex with numerous contributing factors. Computational prediction models often oversimplify this complexity, omitting important considerations, such as inheritance pattern, allelic state, and incomplete penetrance.5 In particular, the phenomenon of incomplete penetrance underscores that not all carriers of a purportedly “pathogenic” allele will necessarily manifest clinical disease. Rather, one's genetic background and environmental exposures can modulate the phenotypic effects of mutations. Fully accounting for such nuances remains a notable challenge for in silico approaches. A more comprehensive integration of diverse data, including family history, genetic context, and environmental interactions, may help advance computational pathogenicity predictions by more closely approximating the intricate interplay of biological and environmental determinants that collectively influence disease risk and expression in the real world.
In summary, as a significantly noteworthy advancement in the field of protein variant analysis, AlphaMissense predicts the possible impact of every amino acid substitution in the human proteome and classifies 89% of missense variants as either likely benign or likely pathogenic. In addition, AlphaMissense interprets the effects of every possible missense mutation in the human genome on protein structure and function based on the insights obtained from protein structure analysis. Overall, by incorporating the pathogenic classification of missense mutations of unknown significance into protein structure-predicting models, AlphaMissense holds promise to identify where disease-causing mutations are likely to occur in a protein.
Ming Yi and Yunqiang Liu drafted the manuscript, Zhiguang Su revised the manuscript. All authors have read and approved the final manuscript.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


