Genome-wide DNA methylation of lesional and peri-lesional skin in vitiligo: a comparative and integrated analysis of multi-omics in Chinese population.
{"title":"Genome-wide DNA methylation of lesional and peri-lesional skin in vitiligo: a comparative and integrated analysis of multi-omics in Chinese population.","authors":"Lin Liu, Yuzhou Xue, Yuxin Li, Yangmei Chen, Xingyu Pan, Yujing Huang, Tingqiao Chen, Judan Zhong, Xinyi Shao, Yihuan Pu, Jin Chen","doi":"10.1007/s00439-023-02630-5","DOIUrl":null,"url":null,"abstract":"<p><p>Several studies have emphasized the role of DNA methylation in vitiligo. However, its profile in human skin of individuals with vitiligo remains unknown. Here, we aimed to study the DNA methylation profile of vitiligo using pairwise comparisons of lesions, peri-lesions, and healthy skin. We investigated DNA methylation levels in six lesional skin, six peri-lesional skin, and eight healthy skin samples using an Illumina 850 K methylation chip. We then integrated DNA methylation data with transcriptome data to identify differentially methylated and expressed genes (DMEGs) and analyzed their functional enrichment. Subsequently, we compared the methylation and transcriptome characteristics of all skin samples, and the related genes were further studied using scRNA-seq data. Finally, validation was performed using an external dataset. We observed more DNA hypomethylated sites in patients with vitiligo. Further integrated analysis identified 264 DMEGs that were mainly functionally enriched in cell division, pigmentation, circadian rhythm, fatty acid metabolism, peroxidase activity, synapse regulation, and extracellular matrix. In addition, in the peri-lesional skin, we found that methylation levels of 102 DMEGs differed prior to changes in their transcription levels and identified 16 key pre-DMEGs (ANLN, CDCA3, CENPA, DEPDC1, ECT2, DEPDC1B, HMMR, KIF18A, KIF18B, TTK, KIF23, DCT, EDNRB, MITF, OCA2, and TYRP1). Single-cell RNA analysis showed that these genes were associated with cycling keratinocytes and melanocytes. Further analysis of cellular communication indicated the involvement of the extracellular matrix. The expression of related genes was verified using an external dataset. To the best of our knowledge, this is the first study to report a comprehensive DNA methylation profile of clinical vitiligo and peri-lesional skin. These findings would contribute to future research on the pathogenesis of vitiligo and potential therapeutic strategies.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"137-149"},"PeriodicalIF":3.6000,"publicationDate":"2024-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02630-5","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
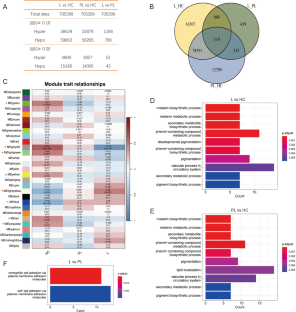
Several studies have emphasized the role of DNA methylation in vitiligo. However, its profile in human skin of individuals with vitiligo remains unknown. Here, we aimed to study the DNA methylation profile of vitiligo using pairwise comparisons of lesions, peri-lesions, and healthy skin. We investigated DNA methylation levels in six lesional skin, six peri-lesional skin, and eight healthy skin samples using an Illumina 850 K methylation chip. We then integrated DNA methylation data with transcriptome data to identify differentially methylated and expressed genes (DMEGs) and analyzed their functional enrichment. Subsequently, we compared the methylation and transcriptome characteristics of all skin samples, and the related genes were further studied using scRNA-seq data. Finally, validation was performed using an external dataset. We observed more DNA hypomethylated sites in patients with vitiligo. Further integrated analysis identified 264 DMEGs that were mainly functionally enriched in cell division, pigmentation, circadian rhythm, fatty acid metabolism, peroxidase activity, synapse regulation, and extracellular matrix. In addition, in the peri-lesional skin, we found that methylation levels of 102 DMEGs differed prior to changes in their transcription levels and identified 16 key pre-DMEGs (ANLN, CDCA3, CENPA, DEPDC1, ECT2, DEPDC1B, HMMR, KIF18A, KIF18B, TTK, KIF23, DCT, EDNRB, MITF, OCA2, and TYRP1). Single-cell RNA analysis showed that these genes were associated with cycling keratinocytes and melanocytes. Further analysis of cellular communication indicated the involvement of the extracellular matrix. The expression of related genes was verified using an external dataset. To the best of our knowledge, this is the first study to report a comprehensive DNA methylation profile of clinical vitiligo and peri-lesional skin. These findings would contribute to future research on the pathogenesis of vitiligo and potential therapeutic strategies.
一些研究强调了 DNA 甲基化在白癜风中的作用。然而,DNA甲基化在白癜风患者皮肤中的分布情况仍不清楚。在这里,我们旨在通过对皮损、皮损周围和健康皮肤进行配对比较来研究白癜风的DNA甲基化概况。我们使用 Illumina 850 K 甲基化芯片研究了六个皮损皮肤、六个皮损周围皮肤和八个健康皮肤样本的 DNA 甲基化水平。然后,我们将 DNA 甲基化数据与转录组数据整合,以确定差异甲基化和表达基因(DMEGs),并分析其功能富集性。随后,我们比较了所有皮肤样本的甲基化和转录组特征,并利用 scRNA-seq 数据进一步研究了相关基因。最后,我们利用外部数据集进行了验证。我们在白癜风患者中观察到了更多的DNA低甲基化位点。进一步的综合分析发现了264个DMEGs,它们主要富集于细胞分裂、色素沉着、昼夜节律、脂肪酸代谢、过氧化物酶活性、突触调节和细胞外基质等方面。此外,我们发现在皮肤周围,102 个 DMEGs 的甲基化水平在其转录水平发生变化之前就存在差异,并确定了 16 个关键的前 DMEGs(ANLN、CDCA3、CENPA、DEPDC1、ECT2、DEPDC1B、HMMR、KIF18A、KIF18B、TTK、KIF23、DCT、EDNRB、MITF、OCA2 和 TYRP1)。单细胞 RNA 分析表明,这些基因与循环角质细胞和黑色素细胞有关。对细胞通讯的进一步分析表明,细胞外基质也参与其中。相关基因的表达已通过外部数据集得到验证。据我们所知,这是第一项报告临床白癜风和皮损周围皮肤 DNA 甲基化概况的研究。这些发现将有助于今后对白癜风发病机制和潜在治疗策略的研究。
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


