{"title":"AmyloComp: A Bioinformatic Tool for Prediction of Amyloid Co-aggregation","authors":"","doi":"10.1016/j.jmb.2024.168437","DOIUrl":null,"url":null,"abstract":"<div><p>Typically, amyloid fibrils consist of multiple copies of the same protein. In these fibrils, each polypeptide chain adopts the same β-arc-containing conformation and these chains are stacked in a parallel and in-register manner. In the last few years, however, a considerable body of data has been accumulated about co-aggregation of different amyloid-forming proteins. Among known examples of the co-aggregation are heteroaggregates of different yeast prions and human proteins Rip1 and Rip3. Since the co-aggregation is linked to such important phenomena as infectivity of amyloids and molecular mechanisms of functional amyloids, we analyzed its structural aspects in more details. An axial stacking of different proteins within the same amyloid fibril is one of the most common type of co-aggregation. By using an approach based on structural similarity of the growing tips of amyloids, we developed a computational method to predict amyloidogenic β-arch structures that are able to interact with each other by the axial stacking. Furthermore, we compiled a dataset consisting of 26 experimentally known pairs of proteins capable or incapable to co-aggregate. We utilized this dataset to test and refine our algorithm. The developed method opens a way for a number of applications, including the identification of microbial proteins capable triggering amyloidosis in humans. AmyloComp is available on the website: <span><span>https://bioinfo.crbm.cnrs.fr/index.php?route=tools&tool=30</span><svg><path></path></svg></span>.</p></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 17","pages":"Article 168437"},"PeriodicalIF":4.7000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0022283624000032/pdfft?md5=7c4b0171bee8cb64ea160d5cea06ba57&pid=1-s2.0-S0022283624000032-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624000032","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
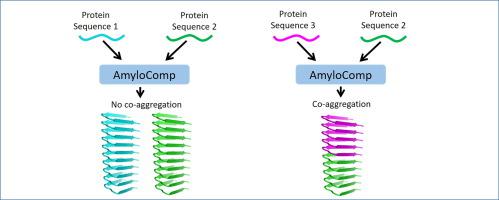
Typically, amyloid fibrils consist of multiple copies of the same protein. In these fibrils, each polypeptide chain adopts the same β-arc-containing conformation and these chains are stacked in a parallel and in-register manner. In the last few years, however, a considerable body of data has been accumulated about co-aggregation of different amyloid-forming proteins. Among known examples of the co-aggregation are heteroaggregates of different yeast prions and human proteins Rip1 and Rip3. Since the co-aggregation is linked to such important phenomena as infectivity of amyloids and molecular mechanisms of functional amyloids, we analyzed its structural aspects in more details. An axial stacking of different proteins within the same amyloid fibril is one of the most common type of co-aggregation. By using an approach based on structural similarity of the growing tips of amyloids, we developed a computational method to predict amyloidogenic β-arch structures that are able to interact with each other by the axial stacking. Furthermore, we compiled a dataset consisting of 26 experimentally known pairs of proteins capable or incapable to co-aggregate. We utilized this dataset to test and refine our algorithm. The developed method opens a way for a number of applications, including the identification of microbial proteins capable triggering amyloidosis in humans. AmyloComp is available on the website: https://bioinfo.crbm.cnrs.fr/index.php?route=tools&tool=30.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


