{"title":"Novel Digenic Variants in <i>COL4A4</i> and <i>COL4A5</i> Causing X-Linked Alport Syndrome: A Case Report.","authors":"Hideki Uedono, Katsuhito Mori, Shinya Nakatani, Kohei Watanabe, Rino Nakaya, Fumiyuki Morioka, Kazuma Sone, Chie Ono, Junko Hotta, Akihiro Tsuda, Naoya Morisada, Toshiyuki Seto, Kandai Nozu, Masanori Emoto","doi":"10.1159/000535493","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Alport syndrome (AS) is a hereditary, progressive kidney disease characterized by structural abnormalities and dysfunction of the glomerular basement membrane (GBM). AS is classified as X-linked, autosomal, and digenic. The number of cases of digenic AS has increased, but the genotype-phenotype correlation of patient with digenic AS is still unclear. Here, we present a case of digenic AS with novel digenic missense variants in <i>COL4A4</i> (c.827G>C, p.Gly276Ala) and <i>COL4A5</i> (c.4369G>C, p.Gly1457Arg).</p><p><strong>Case presentation: </strong>The patient was a 29-year-old Japanese man suffering from persistent microscopic hematuria and proteinuria without kidney function impairment. Kidney biopsy showed focal interstitial foam cell infiltration, global and segmental glomerulosclerosis. Immunofluorescence staining for collagen IV α5 was almost negative in the GBM and Bowman's capsule. Electron microscopy revealed irregular thickening with lamellation and segmental thinning of the GBM. Clinical and pathological findings were consistent with AS. Comprehensive next-generation sequencing revealed a heterozygous missense variant in <i>COL4A4</i> (c.827G>C, p.Gly276Ala) in exon 1 and a hemizygous missense variant in <i>COL4A5</i> (c.4369G>C, p.Gly1457Arg) in exon 49 on the patient's paternal and maternal alleles, respectively. The same digenic variants were detected in his sister, and she also showed a similar phenotype. After treatment with angiotensin-converting enzyme inhibitors, proteinuria decreased from 2.3 to 1.1 g/g creatinine, but occult blood persisted. During follow-up, kidney function has been preserved.</p><p><strong>Conclusion: </strong>The novel genotype of our case provides more information on the genotype-phenotype correlation of digenic XLAS, although long-term follow-up is required. The findings in the present case also indicate the importance of genetic tests for family members of a patient diagnosed with digenic AS.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"14 1","pages":"1-9"},"PeriodicalIF":0.9000,"publicationDate":"2024-01-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10764090/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000535493","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Alport syndrome (AS) is a hereditary, progressive kidney disease characterized by structural abnormalities and dysfunction of the glomerular basement membrane (GBM). AS is classified as X-linked, autosomal, and digenic. The number of cases of digenic AS has increased, but the genotype-phenotype correlation of patient with digenic AS is still unclear. Here, we present a case of digenic AS with novel digenic missense variants in COL4A4 (c.827G>C, p.Gly276Ala) and COL4A5 (c.4369G>C, p.Gly1457Arg).
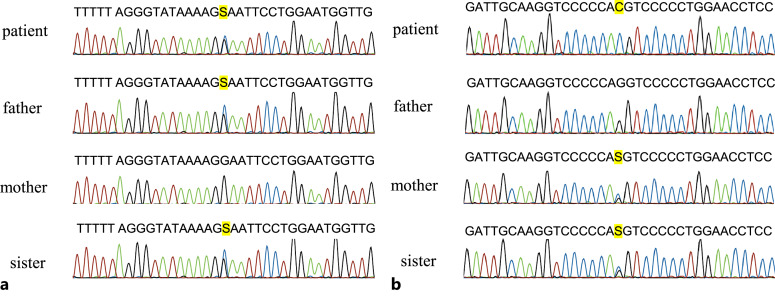
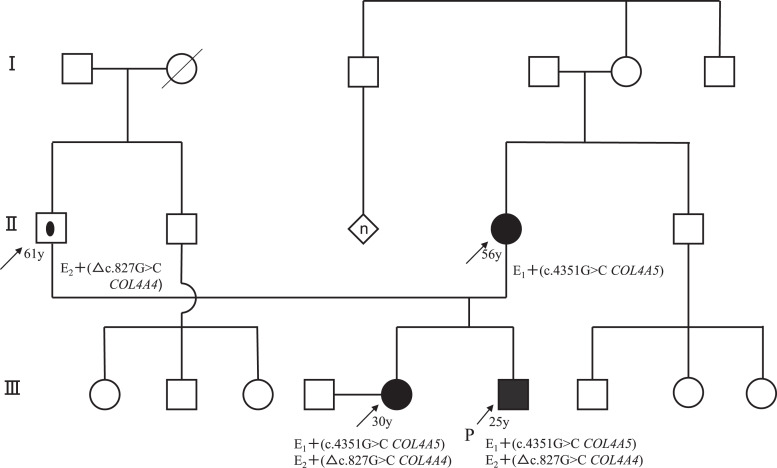
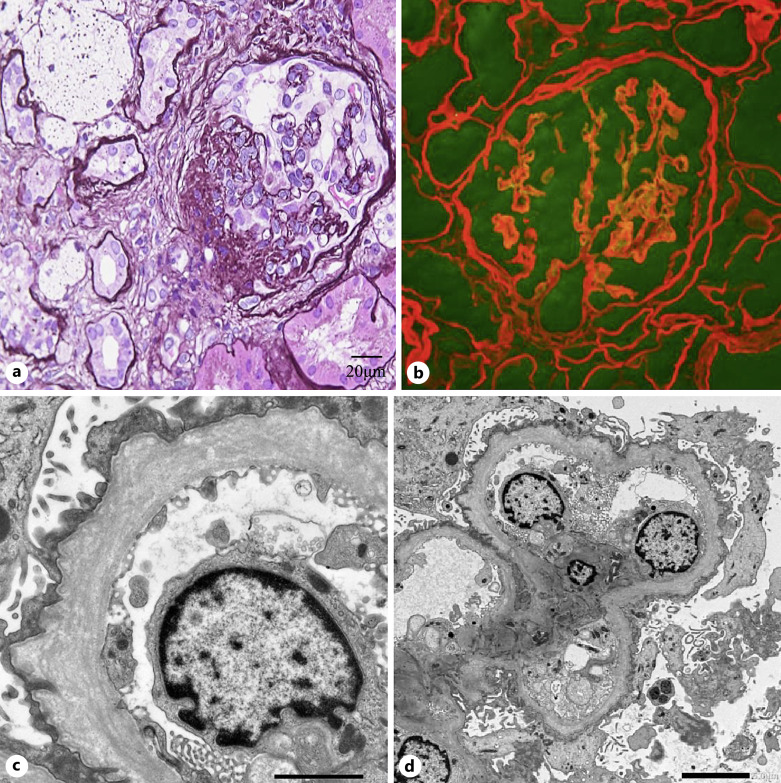
Case presentation: The patient was a 29-year-old Japanese man suffering from persistent microscopic hematuria and proteinuria without kidney function impairment. Kidney biopsy showed focal interstitial foam cell infiltration, global and segmental glomerulosclerosis. Immunofluorescence staining for collagen IV α5 was almost negative in the GBM and Bowman's capsule. Electron microscopy revealed irregular thickening with lamellation and segmental thinning of the GBM. Clinical and pathological findings were consistent with AS. Comprehensive next-generation sequencing revealed a heterozygous missense variant in COL4A4 (c.827G>C, p.Gly276Ala) in exon 1 and a hemizygous missense variant in COL4A5 (c.4369G>C, p.Gly1457Arg) in exon 49 on the patient's paternal and maternal alleles, respectively. The same digenic variants were detected in his sister, and she also showed a similar phenotype. After treatment with angiotensin-converting enzyme inhibitors, proteinuria decreased from 2.3 to 1.1 g/g creatinine, but occult blood persisted. During follow-up, kidney function has been preserved.
Conclusion: The novel genotype of our case provides more information on the genotype-phenotype correlation of digenic XLAS, although long-term follow-up is required. The findings in the present case also indicate the importance of genetic tests for family members of a patient diagnosed with digenic AS.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


