Gokul Srinivasan, Matthew J Davis, Matthew R LeBoeuf, Michael Fatemi, Zarif L Azher, Yunrui Lu, Alos B Diallo, Marietta K Saldias Montivero, Fred W Kolling, Laurent Perrard, Lucas A Salas, Brock C Christensen, Thomas J Palys, Margaret R Karagas, Scott M Palisoul, Gregory J Tsongalis, Louis J Vaickus, Sarah M Preum, Joshua J Levy
{"title":"Potential to Enhance Large Scale Molecular Assessments of Skin Photoaging through Virtual Inference of Spatial Transcriptomics from Routine Staining.","authors":"Gokul Srinivasan, Matthew J Davis, Matthew R LeBoeuf, Michael Fatemi, Zarif L Azher, Yunrui Lu, Alos B Diallo, Marietta K Saldias Montivero, Fred W Kolling, Laurent Perrard, Lucas A Salas, Brock C Christensen, Thomas J Palys, Margaret R Karagas, Scott M Palisoul, Gregory J Tsongalis, Louis J Vaickus, Sarah M Preum, Joshua J Levy","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p>The advent of spatial transcriptomics technologies has heralded a renaissance in research to advance our understanding of the spatial cellular and transcriptional heterogeneity within tissues. Spatial transcriptomics allows investigation of the interplay between cells, molecular pathways, and the surrounding tissue architecture and can help elucidate developmental trajectories, disease pathogenesis, and various niches in the tumor microenvironment. Photoaging is the histological and molecular skin damage resulting from chronic/acute sun exposure and is a major risk factor for skin cancer. Spatial transcriptomics technologies hold promise for improving the reliability of evaluating photoaging and developing new therapeutics. Challenges to current methods include limited focus on dermal elastosis variations and reliance on self-reported measures, which can introduce subjectivity and inconsistency. Spatial transcriptomics offers an opportunity to assess photoaging objectively and reproducibly in studies of carcinogenesis and discern the effectiveness of therapies that intervene in photoaging and preventing cancer. Evaluation of distinct histological architectures using highly-multiplexed spatial technologies can identify specific cell lineages that have been understudied due to their location beyond the depth of UV penetration. However, the cost and interpatient variability using state-of-the-art assays such as the 10x Genomics Spatial Transcriptomics assays limits the scope and scale of large-scale molecular epidemiologic studies. Here, we investigate the inference of spatial transcriptomics information from routine hematoxylin and eosin-stained (H&E) tissue slides. We employed the Visium CytAssist spatial transcriptomics assay to analyze over 18,000 genes at a 50-micron resolution for four patients from a cohort of 261 skin specimens collected adjacent to surgical resection sites for basal cell and squamous cell keratinocyte tumors. The spatial transcriptomics data was co-registered with 40x resolution whole slide imaging (WSI) information. We developed machine learning models that achieved a macro-averaged median AUC and F1 score of 0.80 and 0.61 and Spearman coefficient of 0.60 in inferring transcriptomic profiles across the slides, and accurately captured biological pathways across various tissue architectures.</p>","PeriodicalId":34954,"journal":{"name":"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing","volume":"29 ","pages":"477-491"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10813837/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Computer Science","Score":null,"Total":0}
引用次数: 0
Abstract
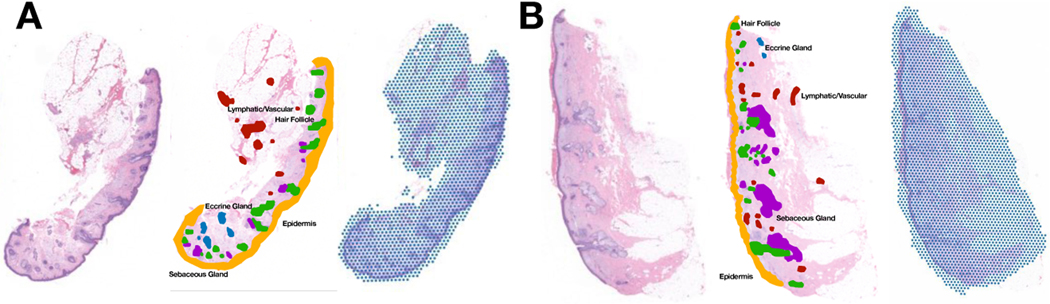
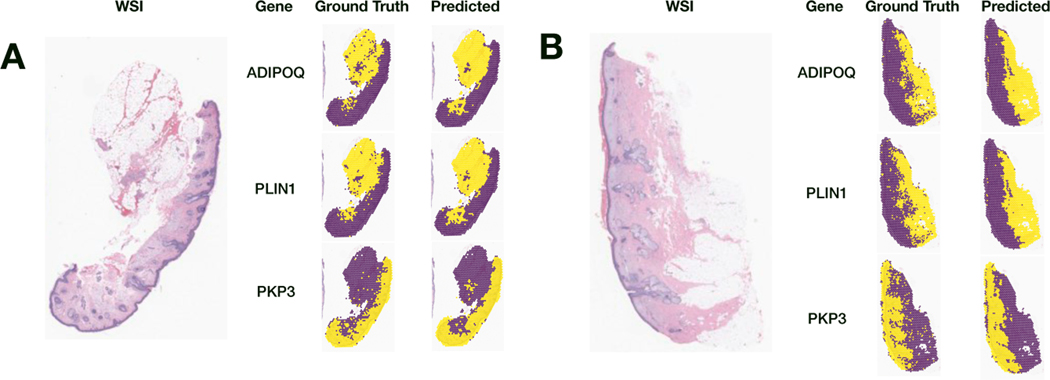
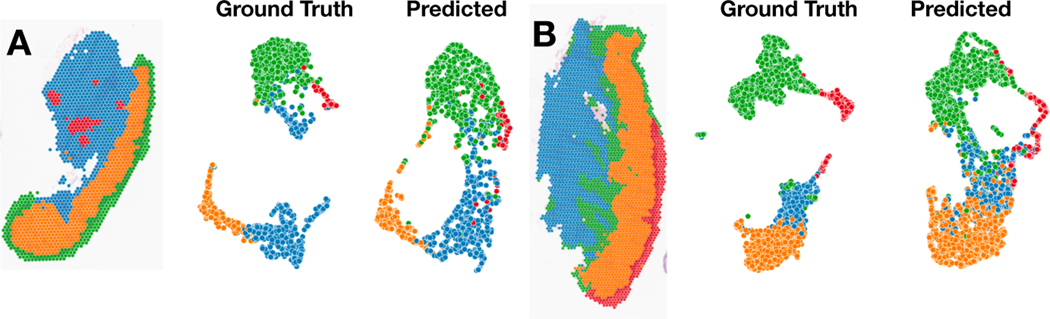
The advent of spatial transcriptomics technologies has heralded a renaissance in research to advance our understanding of the spatial cellular and transcriptional heterogeneity within tissues. Spatial transcriptomics allows investigation of the interplay between cells, molecular pathways, and the surrounding tissue architecture and can help elucidate developmental trajectories, disease pathogenesis, and various niches in the tumor microenvironment. Photoaging is the histological and molecular skin damage resulting from chronic/acute sun exposure and is a major risk factor for skin cancer. Spatial transcriptomics technologies hold promise for improving the reliability of evaluating photoaging and developing new therapeutics. Challenges to current methods include limited focus on dermal elastosis variations and reliance on self-reported measures, which can introduce subjectivity and inconsistency. Spatial transcriptomics offers an opportunity to assess photoaging objectively and reproducibly in studies of carcinogenesis and discern the effectiveness of therapies that intervene in photoaging and preventing cancer. Evaluation of distinct histological architectures using highly-multiplexed spatial technologies can identify specific cell lineages that have been understudied due to their location beyond the depth of UV penetration. However, the cost and interpatient variability using state-of-the-art assays such as the 10x Genomics Spatial Transcriptomics assays limits the scope and scale of large-scale molecular epidemiologic studies. Here, we investigate the inference of spatial transcriptomics information from routine hematoxylin and eosin-stained (H&E) tissue slides. We employed the Visium CytAssist spatial transcriptomics assay to analyze over 18,000 genes at a 50-micron resolution for four patients from a cohort of 261 skin specimens collected adjacent to surgical resection sites for basal cell and squamous cell keratinocyte tumors. The spatial transcriptomics data was co-registered with 40x resolution whole slide imaging (WSI) information. We developed machine learning models that achieved a macro-averaged median AUC and F1 score of 0.80 and 0.61 and Spearman coefficient of 0.60 in inferring transcriptomic profiles across the slides, and accurately captured biological pathways across various tissue architectures.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


