{"title":"Effect of Ge-doping on the adsorption of anions (F−, Cl−, Br−) onto the outer surface of boron nitride nanotube: a DFT study","authors":"Marziyeh Mohammadi, Fahimeh Alirezapour, Azadeh Khanmohammadi","doi":"10.1007/s00214-023-03072-y","DOIUrl":null,"url":null,"abstract":"<p>In this study, the adsorption behavior of anions (F<sup>−</sup>, Cl<sup>−</sup>, Br<sup>−</sup>) on the surface of the single-walled boron nitride nanotube (SWBNNT) is explored using density functional theory (DFT). The interaction between the ions with the pristine BNNT and the Ge-doped BNNT is performed in the solution phase. According to the obtained data, the F<sup>−</sup>@BNNT and F<sup>−</sup>@Ge-BNNT systems have the highest adsorption energy with values of − 329.85 and − 344.71 kJ/mol, respectively. On the other hand, the lowest values have been shown in Cl<sup>−</sup>@BNNT and Cl<sup>−</sup>@Ge-BNNT structures with values of − 31.17 and − 57.79 kJ/mol, respectively. During the complexation, a decrease in the energy gap (E<sub>g</sub>) is accompanied by an increase in the reactivity and electrical conductivity. The HOMO–LUMO energy gaps are found to be the lowest in F<sup>−</sup>@Ge-BNNT with 5.311 eV followed by Cl<sup>−</sup>@Ge-BNNT and Br<sup>−</sup>@Ge-BNNT with 5.299 eV and 5.293 eV, respectively, while these values are 8.028, 8.048, and 7.992 eV for F<sup>−</sup>@BNNT, Cl<sup>−</sup>@BNNT and Br<sup>−</sup>@BNNT, respectively. The intermolecular interactions between the species are also evaluated using the natural bond orbital (NBO) analysis. Finally, to confirm the obtained results, the calculated density of states is depicted.</p>","PeriodicalId":23045,"journal":{"name":"Theoretical Chemistry Accounts","volume":"28 3","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2023-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Chemistry Accounts","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1007/s00214-023-03072-y","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
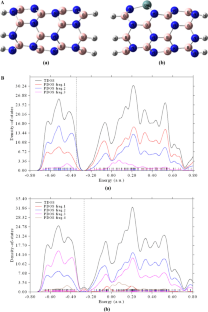
In this study, the adsorption behavior of anions (F−, Cl−, Br−) on the surface of the single-walled boron nitride nanotube (SWBNNT) is explored using density functional theory (DFT). The interaction between the ions with the pristine BNNT and the Ge-doped BNNT is performed in the solution phase. According to the obtained data, the F−@BNNT and F−@Ge-BNNT systems have the highest adsorption energy with values of − 329.85 and − 344.71 kJ/mol, respectively. On the other hand, the lowest values have been shown in Cl−@BNNT and Cl−@Ge-BNNT structures with values of − 31.17 and − 57.79 kJ/mol, respectively. During the complexation, a decrease in the energy gap (Eg) is accompanied by an increase in the reactivity and electrical conductivity. The HOMO–LUMO energy gaps are found to be the lowest in F−@Ge-BNNT with 5.311 eV followed by Cl−@Ge-BNNT and Br−@Ge-BNNT with 5.299 eV and 5.293 eV, respectively, while these values are 8.028, 8.048, and 7.992 eV for F−@BNNT, Cl−@BNNT and Br−@BNNT, respectively. The intermolecular interactions between the species are also evaluated using the natural bond orbital (NBO) analysis. Finally, to confirm the obtained results, the calculated density of states is depicted.
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


