Microbial gene expression analysis of healthy and cancerous esophagus uncovers bacterial biomarkers of clinical outcomes
IF 5.1
Q1 ECOLOGY
引用次数: 0
Abstract
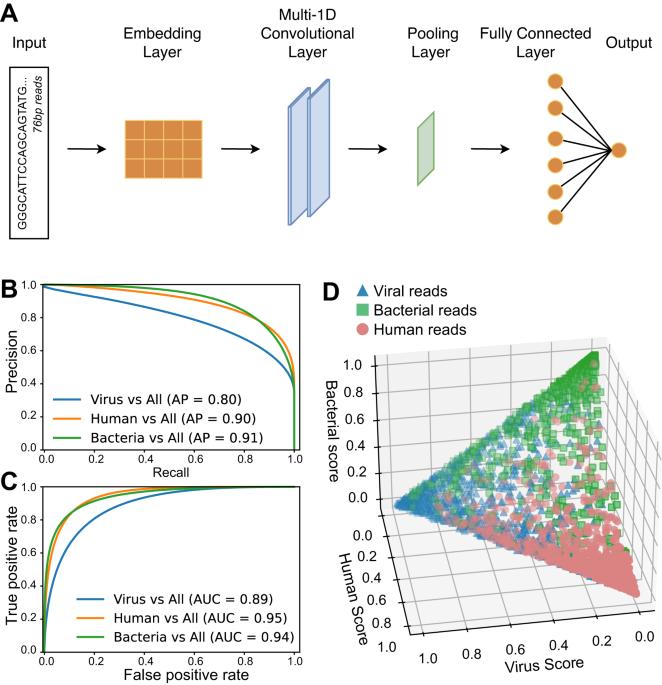
Local microbiome shifts are implicated in the development and progression of gastrointestinal cancers, and in particular, esophageal carcinoma (ESCA), which is among the most aggressive malignancies. Short-read RNA sequencing (RNAseq) is currently the leading technology to study gene expression changes in cancer. However, using RNAseq to study microbial gene expression is challenging. Here, we establish a new tool to efficiently detect viral and bacterial expression in human tissues through RNAseq. This approach employs a neural network to predict reads of likely microbial origin, which are targeted for assembly into longer contigs, improving identification of microbial species and genes. This approach is applied to perform a systematic comparison of bacterial expression in ESCA and healthy esophagi. We uncover bacterial genera that are over or underabundant in ESCA vs healthy esophagi both before and after correction for possible covariates, including patient metadata. However, we find that bacterial taxonomies are not significantly associated with clinical outcomes. Strikingly, in contrast, dozens of microbial proteins were significantly associated with poor patient outcomes and in particular, proteins that perform mitochondrial functions and iron-sulfur coordination. We further demonstrate associations between these microbial proteins and dysregulated host pathways in ESCA patients. Overall, these results suggest possible influences of bacteria on the development of ESCA and uncover new prognostic biomarkers based on microbial genes. In addition, this study provides a framework for the analysis of other human malignancies whose development may be driven by pathogens.
健康和癌性食道的微生物基因表达分析揭示了临床结果的细菌生物标志物。
局部微生物组的变化与胃肠道癌症的发生和进展有关,特别是食管癌(ESCA),这是最具侵袭性的恶性肿瘤之一。短读RNA测序(RNAseq)是目前研究癌症中基因表达变化的领先技术。然而,使用RNAseq来研究微生物基因表达是具有挑战性的。在这里,我们建立了一种新的工具,通过RNAseq有效地检测病毒和细菌在人体组织中的表达。该方法采用神经网络来预测可能的微生物来源的reads,这些reads被组装成更长的contigs,从而提高了微生物物种和基因的识别。该方法应用于ESCA和健康食管中细菌表达的系统比较。我们发现在校正可能的协变量(包括患者元数据)之前和之后,ESCA与健康食管中细菌属过多或过少。然而,我们发现细菌分类与临床结果没有显著相关性。引人注目的是,相比之下,数十种微生物蛋白质与患者预后不良显著相关,特别是执行线粒体功能和铁硫协调的蛋白质。我们进一步证明了这些微生物蛋白与ESCA患者中失调的宿主途径之间的关联。总之,这些结果提示了细菌对ESCA发展的可能影响,并揭示了基于微生物基因的新的预后生物标志物。此外,本研究为分析其他可能由病原体驱动的人类恶性肿瘤提供了一个框架。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


