Modeling Glutaric Aciduria Type I in human neuroblastoma cells recapitulates neuronal damage that can be rescued by gene replacement
IF 4.6
3区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
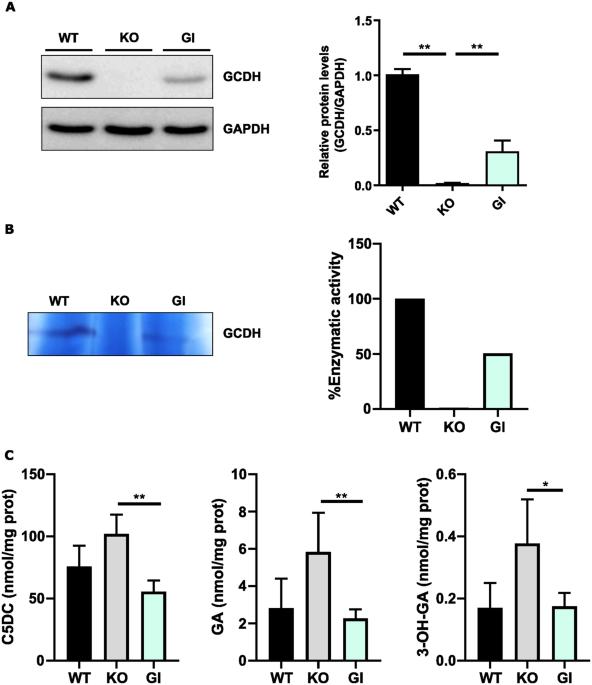
Glutaric Aciduria type I (GA1) is a rare neurometabolic disorder caused by mutations in the GDCH gene encoding for glutaryl-CoA dehydrogenase (GCDH) in the catabolic pathway of lysine, hydroxylysine and tryptophan. GCDH deficiency leads to increased concentrations of glutaric acid (GA) and 3-hydroxyglutaric acid (3-OHGA) in body fluids and tissues. These metabolites are the main triggers of brain damage. Mechanistic studies supporting neurotoxicity in mouse models have been conducted. However, the different vulnerability to some stressors between mouse and human brain cells reveals the need to have a reliable human neuronal model to study GA1 pathogenesis. In the present work we generated a GCDH knockout (KO) in the human neuroblastoma cell line SH-SY5Y by CRISPR/Cas9 technology. SH-SY5Y-GCDH KO cells accumulate GA, 3-OHGA, and glutarylcarnitine when exposed to lysine overload. GA or lysine treatment triggered neuronal damage in GCDH deficient cells. SH-SY5Y-GCDH KO cells also displayed features of GA1 pathogenesis such as increased oxidative stress vulnerability. Restoration of the GCDH activity by gene replacement rescued neuronal alterations. Thus, our findings provide a human neuronal cellular model of GA1 to study this disease and show the potential of gene therapy to rescue GCDH deficiency.
人类神经母细胞瘤细胞戊二酸尿I型模型再现了可通过基因替代拯救的神经元损伤。
戊二酸尿症I型(GA1)是一种罕见的神经代谢性疾病,由编码赖氨酸、羟赖氨酸和色氨酸分解代谢途径中戊二酰辅酶a脱氢酶(GCDH)的GDCH基因突变引起。GCDH缺乏导致体液和组织中戊二酸(GA)和3-羟基戊二酸(3-OHGA)浓度增加。这些代谢物是脑损伤的主要诱因。已经进行了支持小鼠模型神经毒性的机制研究。然而,小鼠和人类脑细胞对某些应激源的易感性不同,表明需要一个可靠的人类神经元模型来研究GA1的发病机制。在目前的工作中,我们通过CRISPR/Cas9技术在人神经母细胞瘤细胞系SH-SY5Y中产生了GCDH敲除(KO)。当暴露于赖氨酸过载时,SH-SY5Y-GCDH KO细胞积累GA, 3-OHGA和戊二酰肉碱。GA或赖氨酸处理可引起GCDH缺陷细胞的神经元损伤。SH-SY5Y-GCDH KO细胞也表现出GA1发病机制的特征,如氧化应激易感性增加。通过基因替代恢复GCDH活性挽救了神经元的改变。因此,我们的发现为研究这种疾病提供了GA1的人类神经元细胞模型,并显示了基因治疗拯救GCDH缺乏症的潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Gene Therapy
医学-生化与分子生物学
CiteScore
9.70
自引率
2.00%
发文量
67
审稿时长
4-8 weeks
期刊介绍:
Gene Therapy covers both the research and clinical applications of novel therapeutic techniques based on a genetic component. Over the last few decades, significant advances in technologies ranging from identifying novel genetic targets that cause disease through to clinical studies, which show therapeutic benefit, have elevated this multidisciplinary field to the forefront of modern medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


