Revealing the influence of tether length on the intramolecular [3 + 2] cycloaddition reactions of nitrones from the molecular electron density theory perspective
{"title":"Revealing the influence of tether length on the intramolecular [3 + 2] cycloaddition reactions of nitrones from the molecular electron density theory perspective","authors":"Asmita Mondal, Luis R. Domingo, Nivedita Acharjee","doi":"10.1002/poc.4574","DOIUrl":null,"url":null,"abstract":"<p>The influence of ethylene substitution and the tether length between the two reacting counterparts on the selectivity and reactivity of the intramolecular [3 + 2] cycloaddition (IM32CA) reactions of cyclic nitrones leading to tricyclic isoxazolidines have been studied within the Molecular Electron Density theory at the MPWB1K/6-311G(d,p) computational level. These <i>zw</i>-type IM32CA reactions follow one-step mechanism, and the activation barrier decreases with the introduction of electron withdrawing (EW) substituent at the alkene moiety in both the intramolecular and intermolecular versions. The IM32CA reactions involving unsubstituted alkene have non-polar character with minimal electron density flux classified as null electron density flux type, while that involving the EW nitro substituted ethylene is more facile with a strong electron density flux from the nitrone to the ethylene moiety, classified as forward electron density flux type. The increase in the polar character of the IM32CA reaction decreases the activation Gibbs free energies associated with these intramolecular processes, while the highly polar IM32CA reactions are disfavored with respect to the intermolecular ones. Interestingly, the preferred regioselectivity observed in low polar IM32CA reactions having three methylene units between the nitrone and ethylene frameworks is reversed to that in nitrones separated with four methylene units, in conformity with the experimental outcome. Finally, electron localization function and quantum theory of atoms-in-molecules studies reveal that, in general, these IM32CA reactions involve early transition state structures in which the formation of new C-C and C-O single bonds have not yet started.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":null,"pages":null},"PeriodicalIF":1.9000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4574","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
Abstract
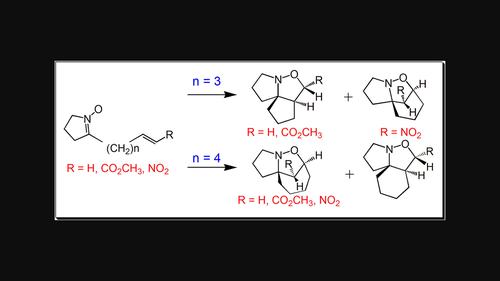
The influence of ethylene substitution and the tether length between the two reacting counterparts on the selectivity and reactivity of the intramolecular [3 + 2] cycloaddition (IM32CA) reactions of cyclic nitrones leading to tricyclic isoxazolidines have been studied within the Molecular Electron Density theory at the MPWB1K/6-311G(d,p) computational level. These zw-type IM32CA reactions follow one-step mechanism, and the activation barrier decreases with the introduction of electron withdrawing (EW) substituent at the alkene moiety in both the intramolecular and intermolecular versions. The IM32CA reactions involving unsubstituted alkene have non-polar character with minimal electron density flux classified as null electron density flux type, while that involving the EW nitro substituted ethylene is more facile with a strong electron density flux from the nitrone to the ethylene moiety, classified as forward electron density flux type. The increase in the polar character of the IM32CA reaction decreases the activation Gibbs free energies associated with these intramolecular processes, while the highly polar IM32CA reactions are disfavored with respect to the intermolecular ones. Interestingly, the preferred regioselectivity observed in low polar IM32CA reactions having three methylene units between the nitrone and ethylene frameworks is reversed to that in nitrones separated with four methylene units, in conformity with the experimental outcome. Finally, electron localization function and quantum theory of atoms-in-molecules studies reveal that, in general, these IM32CA reactions involve early transition state structures in which the formation of new C-C and C-O single bonds have not yet started.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


