Vibronic dynamics from real-time time-dependent density-functional theory coupled to the Ehrenfest scheme: the example of p-coumaric acid
IF 1.5
4区 化学
Q4 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Abstract We investigate the vibronic dynamics of a modified version of the p-coumaric acid using real-time time-dependent density-functional theory coupled with the Ehrenfest scheme in the adiabatic local density approximation. Due to the issues of this functional to yield a reliable starting point for the evolution of the electron-nuclear system triggered by a pulse, we start off the simulations constraining the electronic occupation of the molecule in two excited states corresponding to a bright, delocalized transition, and a dark, charge-transfer-like excitation. By monitoring the kinetic energy spectral density, we analyze the nature of the nuclear motion over a time window of 300 fs. Anharmonic effects appear at low frequencies, below 500 cm $$^{-1}$$
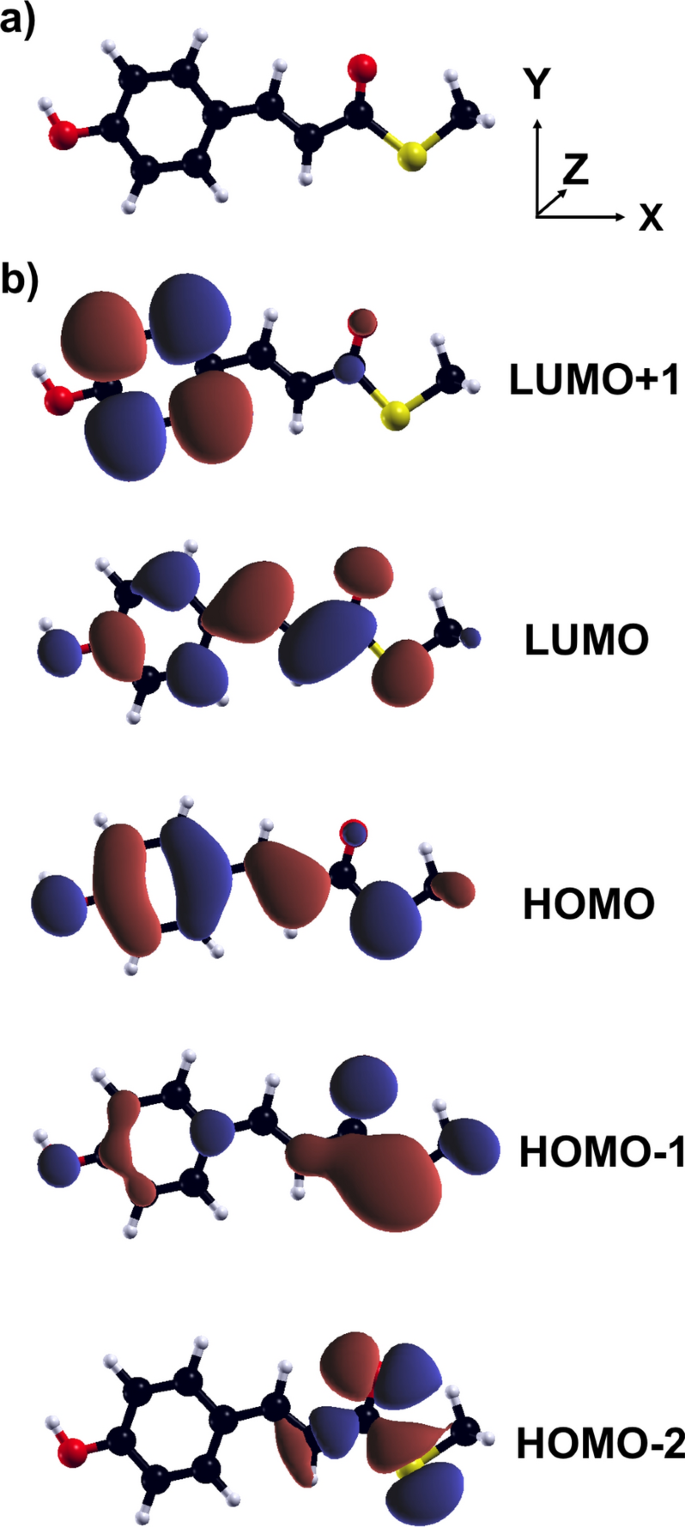
从实时时变密度泛函理论耦合到Ehrenfest方案的振动动力学:以对香豆酸为例
摘要利用实时时变密度泛函理论和绝热局部密度近似中的Ehrenfest格式,研究了一种改进的对香豆酸的振动动力学。由于这一功能的问题,以产生一个可靠的起点为演化的电子-核系统的脉冲触发,我们开始模拟限制电子占据分子在两个激发态对应于一个明亮的,离域跃迁,和一个黑暗的,电荷转移样激发。通过监测动能谱密度,我们分析了300 fs时间窗内核运动的性质。非谐波效应出现在低频率,低于500厘米$$^{-1}$$ - 1,并在电荷转移激发特别明显。在这种情况下,在大约200fs后,分子骨架变得很大程度上扭曲,最初受约束的占据演变成不同的电子构型。另一方面,由离域亮激发初始化的动力学在电子和结构上是稳定的,由此产生的核运动明显是谐波的。我们的研究结果为破译这种发色团和相关系统的振动动力学提供了指示,因为在现实环境中嵌入分子的更详细的模拟。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Theoretical Chemistry Accounts
化学-物理化学
CiteScore
3.40
自引率
0.00%
发文量
74
审稿时长
3.8 months
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


