Thomas Bondo Pedersen, Susi Lehtola, Ignacio Fdez. Galván, Roland Lindh
{"title":"The versatility of the Cholesky decomposition in electronic structure theory","authors":"Thomas Bondo Pedersen, Susi Lehtola, Ignacio Fdez. Galván, Roland Lindh","doi":"10.1002/wcms.1692","DOIUrl":null,"url":null,"abstract":"<p>The resolution-of-the-identity (RI) or density fitting (DF) approximation for the electron repulsion integrals (ERIs) has become a standard component of accelerated and reduced-scaling implementations of first-principles Gaussian-type orbital electronic-structure methods. The Cholesky decomposition (CD) of the ERIs has also become increasingly deployed across quantum chemistry packages in the last decade, even though its early applications were mostly limited to high-accuracy methods such as coupled-cluster theory and multiconfigurational approaches. Starting with a summary of the basic theory underpinning both the CD and RI/DF approximations, thus underlining the extremely close relation of the CD and RI/DF techniques, we provide a brief and largely chronological review of the evolution of the CD approach from its birth in 1977 to its current state. In addition to being a purely numerical procedure for handling ERIs, thus providing robust and computationally efficient approximations to the exact ERIs that have been found increasingly useful on modern computer platforms, CD also offers highly accurate approaches for generating auxiliary basis sets for the RI/DF approximation on the fly due to the deep mathematical connection between the two approaches. In this review, we aim to provide a concise reference of the main techniques employed in various CD approaches in electronic structure theory, to exemplify the connection between the CD and RI/DF approaches, and to clarify the state of the art to guide new implementations of CD approaches across electronic structure programs.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2023-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1692","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1692","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
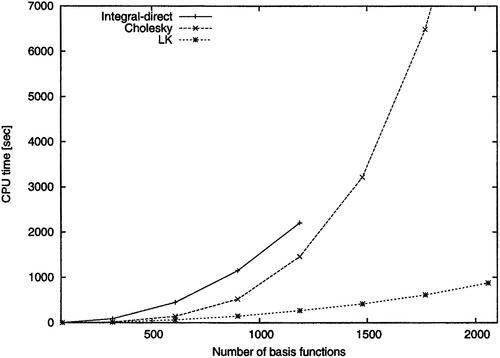
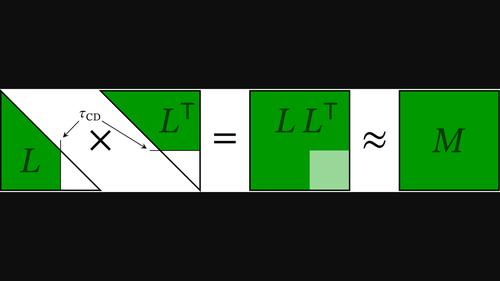
The resolution-of-the-identity (RI) or density fitting (DF) approximation for the electron repulsion integrals (ERIs) has become a standard component of accelerated and reduced-scaling implementations of first-principles Gaussian-type orbital electronic-structure methods. The Cholesky decomposition (CD) of the ERIs has also become increasingly deployed across quantum chemistry packages in the last decade, even though its early applications were mostly limited to high-accuracy methods such as coupled-cluster theory and multiconfigurational approaches. Starting with a summary of the basic theory underpinning both the CD and RI/DF approximations, thus underlining the extremely close relation of the CD and RI/DF techniques, we provide a brief and largely chronological review of the evolution of the CD approach from its birth in 1977 to its current state. In addition to being a purely numerical procedure for handling ERIs, thus providing robust and computationally efficient approximations to the exact ERIs that have been found increasingly useful on modern computer platforms, CD also offers highly accurate approaches for generating auxiliary basis sets for the RI/DF approximation on the fly due to the deep mathematical connection between the two approaches. In this review, we aim to provide a concise reference of the main techniques employed in various CD approaches in electronic structure theory, to exemplify the connection between the CD and RI/DF approaches, and to clarify the state of the art to guide new implementations of CD approaches across electronic structure programs.
电子斥力积分(ERIs)的同一性解析(RI)或密度拟合(DF)近似已成为第一原理高斯轨道电子结构方法加速和缩减缩放实施的标准组成部分。ERIs的Cholesky分解(CD)在过去十年中也越来越多地应用于量子化学软件包中,尽管其早期应用主要局限于高精度方法,如耦合簇理论和多配置方法。我们首先总结了 CD 和 RI/DF 近似的基础理论,从而强调了 CD 和 RI/DF 技术之间极为密切的关系,然后按时间顺序简要回顾了 CD 方法从 1977 年诞生到现在的演变过程。CD 是一种处理 ERI 的纯数值程序,可为精确 ERI 提供稳健且计算效率高的近似值,在现代计算机平台上越来越有用;此外,由于 RI/DF 近似与 CD 两种方法之间存在深层数学联系,CD 还可为 RI/DF 近似提供高精度的辅助基集生成方法。在这篇综述中,我们旨在简明扼要地介绍电子结构理论中各种 CD 方法所采用的主要技术,举例说明 CD 和 RI/DF 方法之间的联系,并阐明目前的技术水平,以指导电子结构程序中 CD 方法的新实施:
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


