{"title":"Machine learning prediction of self-assembly and analysis of molecular structure dependence on the critical packing parameter","authors":"Yuuki Ishiwatari, Takahiro Yokoyama, Tomoya Kojima, Taisuke Banno and Noriyoshi Arai","doi":"10.1039/D3ME00151B","DOIUrl":null,"url":null,"abstract":"<p >Amphiphilic molecules spontaneously form self-assembly structures depending on physical conditions such as the molecular structure, concentration, and temperature. These structures exhibit various functionalities according to their morphology. The critical packing parameter (CPP) is used to correlate self-organized structures with the chemical composition. However, accurately calculating it requires information about both the molecular shape and molecular aggregates, making it challenging to apply directly in molecular design. We aimed to predict the self-assembled structure of a molecule directly from its chemical structure and to analyze the factors influencing it using machine learning. Dissipative particle dynamics simulations were used to reproduce many self-assembly structures comprising various chemical structures, and their CPPs were calculated. Machine learning models were built using the chemical structures as input data and the CPPs as output data. As a result, both random forest and the gated recurrent unit showed high prediction accuracy. Feature importance analysis and sample size dependence revealed that the amphiphilic nature of molecules significantly influences the self-assembly structures. Additionally, selecting an appropriate molecular structure representation for each algorithm is crucial. The study results should contribute to product development in the fields of materials science, materials chemistry, and medical materials.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 1","pages":" 20-28"},"PeriodicalIF":3.2000,"publicationDate":"2023-10-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/me/d3me00151b?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d3me00151b","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
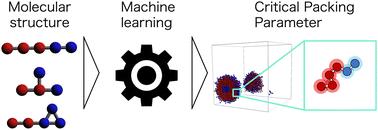
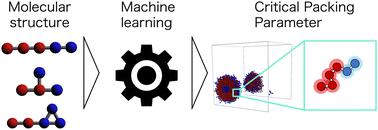
Amphiphilic molecules spontaneously form self-assembly structures depending on physical conditions such as the molecular structure, concentration, and temperature. These structures exhibit various functionalities according to their morphology. The critical packing parameter (CPP) is used to correlate self-organized structures with the chemical composition. However, accurately calculating it requires information about both the molecular shape and molecular aggregates, making it challenging to apply directly in molecular design. We aimed to predict the self-assembled structure of a molecule directly from its chemical structure and to analyze the factors influencing it using machine learning. Dissipative particle dynamics simulations were used to reproduce many self-assembly structures comprising various chemical structures, and their CPPs were calculated. Machine learning models were built using the chemical structures as input data and the CPPs as output data. As a result, both random forest and the gated recurrent unit showed high prediction accuracy. Feature importance analysis and sample size dependence revealed that the amphiphilic nature of molecules significantly influences the self-assembly structures. Additionally, selecting an appropriate molecular structure representation for each algorithm is crucial. The study results should contribute to product development in the fields of materials science, materials chemistry, and medical materials.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


