Liwei Chang, Arup Mondal, Bhumika Singh, Yisel Martínez-Noa, Alberto Perez
{"title":"Revolutionizing peptide-based drug discovery: Advances in the post-AlphaFold era","authors":"Liwei Chang, Arup Mondal, Bhumika Singh, Yisel Martínez-Noa, Alberto Perez","doi":"10.1002/wcms.1693","DOIUrl":null,"url":null,"abstract":"<p>Peptide-based drugs offer high specificity, potency, and selectivity. However, their inherent flexibility and differences in conformational preferences between their free and bound states create unique challenges that have hindered progress in effective drug discovery pipelines. The emergence of AlphaFold (AF) and Artificial Intelligence (AI) presents new opportunities for enhancing peptide-based drug discovery. We explore recent advancements that facilitate a successful peptide drug discovery pipeline, considering peptides' attractive therapeutic properties and strategies to enhance their stability and bioavailability. AF enables efficient and accurate prediction of peptide-protein structures, addressing a critical requirement in computational drug discovery pipelines. In the post-AF era, we are witnessing rapid progress with the potential to revolutionize peptide-based drug discovery such as the ability to rank peptide binders or classify them as binders/non-binders and the ability to design novel peptide sequences. However, AI-based methods are struggling due to the lack of well-curated datasets, for example to accommodate modified amino acids or unconventional cyclization. Thus, physics-based methods, such as docking or molecular dynamics simulations, continue to hold a complementary role in peptide drug discovery pipelines. Moreover, MD-based tools offer valuable insights into binding mechanisms, as well as the thermodynamic and kinetic properties of complexes. As we navigate this evolving landscape, a synergistic integration of AI and physics-based methods holds the promise of reshaping the landscape of peptide-based drug discovery.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2023-11-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1693","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
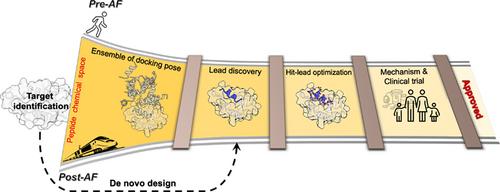
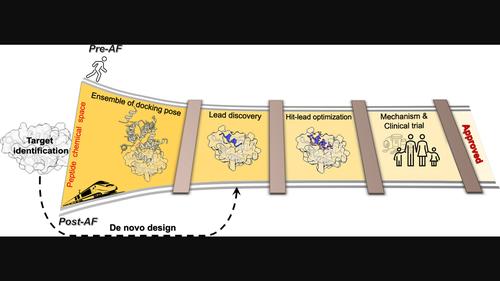
Peptide-based drugs offer high specificity, potency, and selectivity. However, their inherent flexibility and differences in conformational preferences between their free and bound states create unique challenges that have hindered progress in effective drug discovery pipelines. The emergence of AlphaFold (AF) and Artificial Intelligence (AI) presents new opportunities for enhancing peptide-based drug discovery. We explore recent advancements that facilitate a successful peptide drug discovery pipeline, considering peptides' attractive therapeutic properties and strategies to enhance their stability and bioavailability. AF enables efficient and accurate prediction of peptide-protein structures, addressing a critical requirement in computational drug discovery pipelines. In the post-AF era, we are witnessing rapid progress with the potential to revolutionize peptide-based drug discovery such as the ability to rank peptide binders or classify them as binders/non-binders and the ability to design novel peptide sequences. However, AI-based methods are struggling due to the lack of well-curated datasets, for example to accommodate modified amino acids or unconventional cyclization. Thus, physics-based methods, such as docking or molecular dynamics simulations, continue to hold a complementary role in peptide drug discovery pipelines. Moreover, MD-based tools offer valuable insights into binding mechanisms, as well as the thermodynamic and kinetic properties of complexes. As we navigate this evolving landscape, a synergistic integration of AI and physics-based methods holds the promise of reshaping the landscape of peptide-based drug discovery.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


