{"title":"Ring kinematics-informed conformation space exploration","authors":"Nikolai V. Krivoshchapov, Michael G. Medvedev","doi":"10.1002/wcms.1690","DOIUrl":null,"url":null,"abstract":"<p>Conformational searches and ML-driven geometry predictions (e.g., AlphaFold) work in the space of molecule's degrees of freedom. When dealing with cycles, cyclicity constraints impose complex interdependence between them, so that arbitrary changes of cyclic dihedral angles lead to heavy distortions of some bond lengths and valence angles of the ring. This renders navigation through conformational space of cyclic molecules to be very challenging. Inverse kinematics is a theory that provides a mathematically strict solution to this problem. It allows one to identify degrees of freedom for any polycyclic molecule, that is, its dihedral angles that can be set independently from each other. Then for any values of degrees of freedom, inverse kinematics can reconstruct the remaining dihedrals so that all rings are closed with given bond lengths and valence angles. This approach offers a handy and efficient way for constructing and navigating conformational space of any molecule using either naïve Monte-Carlo sampling or sophisticated machine learning models. Inverse kinematics can considerably narrow the conformational space of a polycyclic molecule to include only cyclicity-preserving regions. Thus, it can be viewed as a physical constraint on the model, making the latter obey the laws of kinematics, which govern the rings conformations. We believe that inverse kinematics will be universally used in the ever-growing field of geometry prediction of complex interlinked molecules.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2023-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1690","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
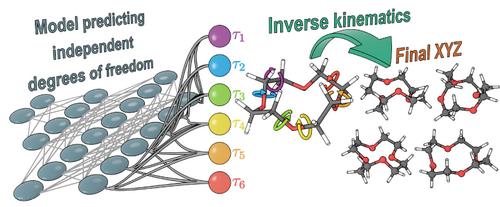
Abstract
Conformational searches and ML-driven geometry predictions (e.g., AlphaFold) work in the space of molecule's degrees of freedom. When dealing with cycles, cyclicity constraints impose complex interdependence between them, so that arbitrary changes of cyclic dihedral angles lead to heavy distortions of some bond lengths and valence angles of the ring. This renders navigation through conformational space of cyclic molecules to be very challenging. Inverse kinematics is a theory that provides a mathematically strict solution to this problem. It allows one to identify degrees of freedom for any polycyclic molecule, that is, its dihedral angles that can be set independently from each other. Then for any values of degrees of freedom, inverse kinematics can reconstruct the remaining dihedrals so that all rings are closed with given bond lengths and valence angles. This approach offers a handy and efficient way for constructing and navigating conformational space of any molecule using either naïve Monte-Carlo sampling or sophisticated machine learning models. Inverse kinematics can considerably narrow the conformational space of a polycyclic molecule to include only cyclicity-preserving regions. Thus, it can be viewed as a physical constraint on the model, making the latter obey the laws of kinematics, which govern the rings conformations. We believe that inverse kinematics will be universally used in the ever-growing field of geometry prediction of complex interlinked molecules.
构象搜索和 ML 驱动的几何预测(如 AlphaFold)是在分子自由度空间中进行的。在处理循环时,循环约束在它们之间施加了复杂的相互依存关系,因此任意改变循环二面角会导致环的某些键长和价角发生严重扭曲。这使得在环状分子的构象空间中进行导航非常具有挑战性。逆运动学理论为这一问题提供了严格的数学解决方案。它允许我们确定任何多环分子的自由度,即可以独立设置的二面角。然后,对于任何自由度值,逆运动学都可以重建剩余的二面角,从而使所有环都以给定的键长和价角闭合。这种方法提供了一种方便、高效的方法,可以使用天真的蒙特卡洛采样或复杂的机器学习模型来构建和浏览任何分子的构象空间。逆运动学可以大大缩小多环分子的构象空间,使其只包括保留环性的区域。因此,它可以被视为对模型的一种物理约束,使后者遵守运动学定律,从而控制环的构象。我们相信,逆运动学将被广泛应用于日益增长的复杂互联分子几何预测领域:
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


