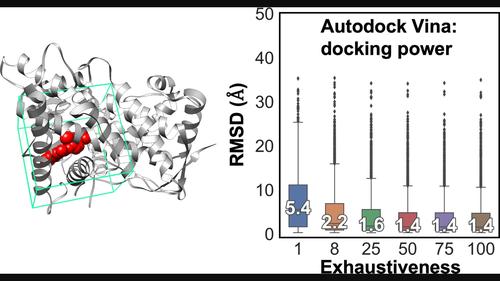
{"title":"Speed vs Accuracy: Effect on Ligand Pose Accuracy of Varying Box Size and Exhaustiveness in AutoDock Vina.","authors":"Rupesh Agarwal, Jeremy C Smith","doi":"10.1002/minf.202200188","DOIUrl":null,"url":null,"abstract":"<p><p>Structure-based virtual high-throughput screening involves docking chemical libraries to targets of interest. A parameter pertinent to the accuracy of the resulting pose is the root mean square deviation (RMSD) from a known crystallographic structure, i. e., the 'docking power'. Here, using a popular algorithm, Autodock Vina, as a model program, we evaluate the effects of varying two common docking parameters: the box size (the size of docking search space) and the exhaustiveness of the global search (the number of independent runs starting from random ligand conformations) on the RMSD from the PDBbind v2017 refined dataset of experimental protein-ligand complexes. Although it is clear that exhaustiveness is an important parameter, there is wide variation in the values used, with variation between 1 and >100. We, therefore, evaluated a combination of cubic boxes of different sizes and five exhaustiveness values (1, 8, 25, 50, 75, 100) within the range of those commonly adopted. The results show that the default exhaustiveness value of 8 performs well overall for most box sizes. In contrast, for all box sizes, but particularly for large boxes, an exhaustiveness value of 1 led to significantly higher median RMSD (mRMSD) values. The docking power was slightly improved with an exhaustiveness of 25, but the mRMSD changes little with values higher than 25. Therefore, although low exhaustiveness is computationally faster, the results are more likely to be far from reality, and, conversely, values >25 led to little improvement at the expense of computational resources. Overall, we recommend users to use at least the default exhaustiveness value of 8 for virtual screening calculations.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":null,"pages":null},"PeriodicalIF":2.8000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"13","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200188","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 13
Abstract
Structure-based virtual high-throughput screening involves docking chemical libraries to targets of interest. A parameter pertinent to the accuracy of the resulting pose is the root mean square deviation (RMSD) from a known crystallographic structure, i. e., the 'docking power'. Here, using a popular algorithm, Autodock Vina, as a model program, we evaluate the effects of varying two common docking parameters: the box size (the size of docking search space) and the exhaustiveness of the global search (the number of independent runs starting from random ligand conformations) on the RMSD from the PDBbind v2017 refined dataset of experimental protein-ligand complexes. Although it is clear that exhaustiveness is an important parameter, there is wide variation in the values used, with variation between 1 and >100. We, therefore, evaluated a combination of cubic boxes of different sizes and five exhaustiveness values (1, 8, 25, 50, 75, 100) within the range of those commonly adopted. The results show that the default exhaustiveness value of 8 performs well overall for most box sizes. In contrast, for all box sizes, but particularly for large boxes, an exhaustiveness value of 1 led to significantly higher median RMSD (mRMSD) values. The docking power was slightly improved with an exhaustiveness of 25, but the mRMSD changes little with values higher than 25. Therefore, although low exhaustiveness is computationally faster, the results are more likely to be far from reality, and, conversely, values >25 led to little improvement at the expense of computational resources. Overall, we recommend users to use at least the default exhaustiveness value of 8 for virtual screening calculations.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


