{"title":"Tumor necroptosis-mediated shedding of cell surface proteins promotes metastasis of breast cancer by suppressing anti-tumor immunity.","authors":"Zhaoshan Liu, Swati Choksi, Hyung-Joon Kwon, Delong Jiao, Chengyu Liu, Zheng-Gang Liu","doi":"10.1186/s13058-023-01604-9","DOIUrl":null,"url":null,"abstract":"<p><p>Necroptosis is a form of regulated necrosis and is executed by MLKL when MLKL is engaged in triggering the rupture of cell plasma membrane. MLKL activation also leads to the protease, ADAMs-mediated ectodomain shedding of cell surface proteins of necroptotic cells. Tumor necroptosis often happens in advanced solid tumors, and blocking necroptosis by MLKL deletion in breast cancer dramatically reduces tumor metastasis. It has been suggested that tumor necroptosis affects tumor progression through modulating the tumor microenvironment. However, the exact mechanism by which tumor necroptosis promotes tumor metastasis remains elusive. Here, we report that the ectodomain shedding of cell surface proteins of necroptotic cells is critical for the promoting effect of tumor necroptosis in tumor metastasis through inhibiting the anti-tumor activity of T cells. We found that blocking tumor necroptosis by MLKL deletion led to the dramatic reduction of tumor metastasis and significantly elevated anti-tumor activity of tumor-infiltrating and peripheral blood T cells. Importantly, the increased anti-tumor activity of T cells is a key cause for the reduced metastasis as the depletion of CD8+ T cells completely restored the level of metastasis in the Mlkl KO mice. Interestingly, the levels of some soluble cell surface proteins including sE-cadherin that are known to promote metastasis are also dramatically reduced in MLKL null tumors/mice. Administration of ADAMs pan inhibitor reduces the levels of soluble cell surface proteins in WT tumors/mice and leads to the dramatic decrease in metastasis. Finally, we showed the sE-cadherin/KLRG1 inhibitory receptor is the major pathway for necroptosis-mediated suppression of the anti-tumor activity of T cells and the promotion of metastasis. Hence, our study reveals a novel mechanism of tumor necroptosis-mediated promotion of metastasis and suggests that tumor necroptosis and necroptosis-activated ADAMs are potential targets for controlling metastasis.</p>","PeriodicalId":9283,"journal":{"name":"Breast Cancer Research : BCR","volume":"25 1","pages":"10"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9881343/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Breast Cancer Research : BCR","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13058-023-01604-9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 4
Abstract
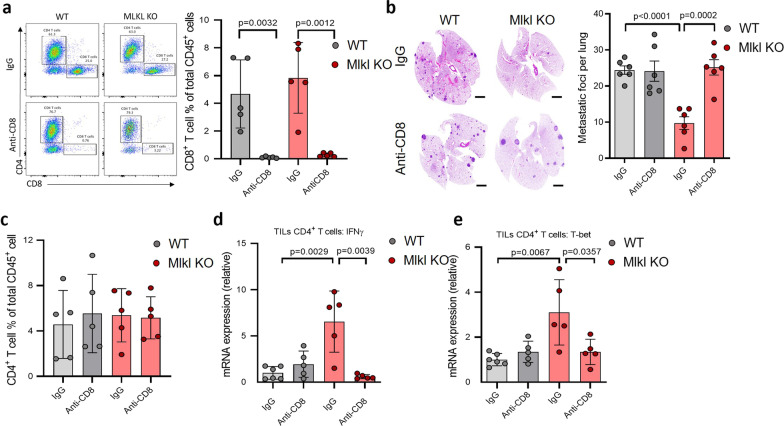
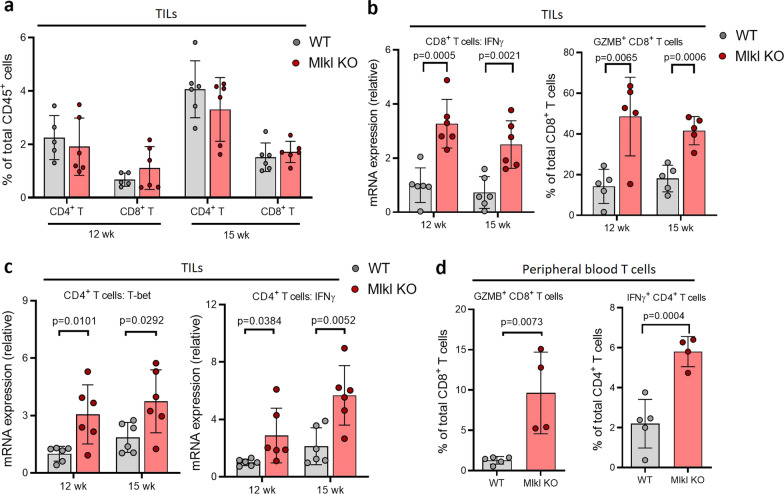
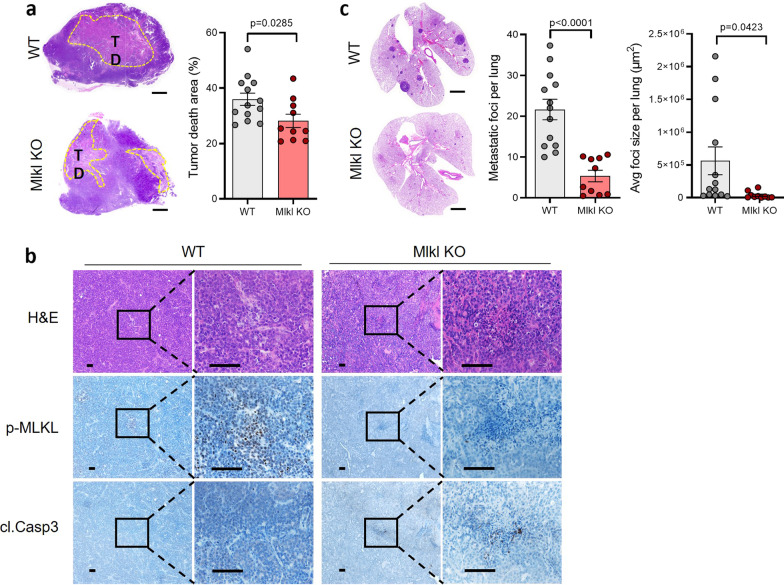
Necroptosis is a form of regulated necrosis and is executed by MLKL when MLKL is engaged in triggering the rupture of cell plasma membrane. MLKL activation also leads to the protease, ADAMs-mediated ectodomain shedding of cell surface proteins of necroptotic cells. Tumor necroptosis often happens in advanced solid tumors, and blocking necroptosis by MLKL deletion in breast cancer dramatically reduces tumor metastasis. It has been suggested that tumor necroptosis affects tumor progression through modulating the tumor microenvironment. However, the exact mechanism by which tumor necroptosis promotes tumor metastasis remains elusive. Here, we report that the ectodomain shedding of cell surface proteins of necroptotic cells is critical for the promoting effect of tumor necroptosis in tumor metastasis through inhibiting the anti-tumor activity of T cells. We found that blocking tumor necroptosis by MLKL deletion led to the dramatic reduction of tumor metastasis and significantly elevated anti-tumor activity of tumor-infiltrating and peripheral blood T cells. Importantly, the increased anti-tumor activity of T cells is a key cause for the reduced metastasis as the depletion of CD8+ T cells completely restored the level of metastasis in the Mlkl KO mice. Interestingly, the levels of some soluble cell surface proteins including sE-cadherin that are known to promote metastasis are also dramatically reduced in MLKL null tumors/mice. Administration of ADAMs pan inhibitor reduces the levels of soluble cell surface proteins in WT tumors/mice and leads to the dramatic decrease in metastasis. Finally, we showed the sE-cadherin/KLRG1 inhibitory receptor is the major pathway for necroptosis-mediated suppression of the anti-tumor activity of T cells and the promotion of metastasis. Hence, our study reveals a novel mechanism of tumor necroptosis-mediated promotion of metastasis and suggests that tumor necroptosis and necroptosis-activated ADAMs are potential targets for controlling metastasis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


