{"title":"A Q-learning method based on coarse-to-fine potential energy surface for locating transition state and reaction pathway","authors":"Wenjun Xu, Yanling Zhao, Jialu Chen, Zhongyu Wan, Dadong Yan, Xinghua Zhang, Ruiqin Zhang","doi":"10.1002/jcc.27259","DOIUrl":null,"url":null,"abstract":"<p>Transition state (TS) on the potential energy surface (PES) plays a key role in determining the kinetics and thermodynamics of chemical reactions. Inspired by the fact that the dynamics of complex systems are always driven by rare but significant transition events, we herein propose a TS search method in accordance with the Q-learning algorithm. Appropriate reward functions are set for a given PES to optimize the reaction pathway through continuous trial and error, and then the TS can be obtained from the optimized reaction pathway. The validity of this Q-learning method with reasonable settings of <i>Q</i>-value table including actions, states, learning rate, greedy rate, discount rate, and so on, is exemplified in 2 two-dimensional potential functions. In the applications of the Q-learning method to two chemical reactions, it is demonstrated that the Q-learning method can predict consistent TS and reaction pathway with those by ab initio calculations. Notably, the PES must be well prepared before using the Q-learning method, and a coarse-to-fine PES scanning scheme is thus introduced to save the computational time while maintaining the accuracy of the Q-learning prediction. This work offers a simple and reliable Q-learning method to search for all possible TS and reaction pathway of a chemical reaction, which may be a new option for effectively exploring the PES in an extensive search manner.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 8","pages":"487-497"},"PeriodicalIF":3.4000,"publicationDate":"2023-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27259","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
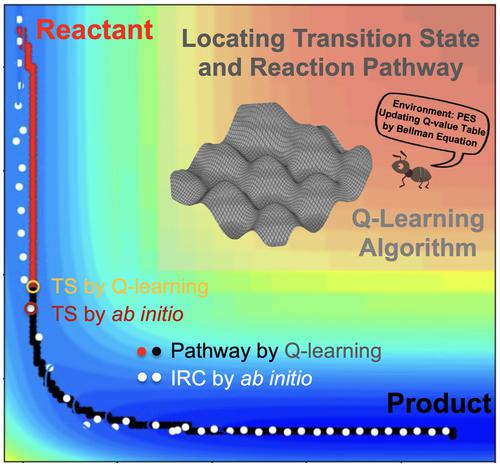
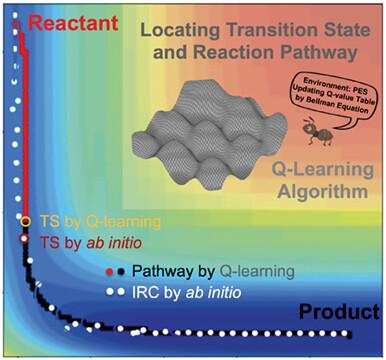
Transition state (TS) on the potential energy surface (PES) plays a key role in determining the kinetics and thermodynamics of chemical reactions. Inspired by the fact that the dynamics of complex systems are always driven by rare but significant transition events, we herein propose a TS search method in accordance with the Q-learning algorithm. Appropriate reward functions are set for a given PES to optimize the reaction pathway through continuous trial and error, and then the TS can be obtained from the optimized reaction pathway. The validity of this Q-learning method with reasonable settings of Q-value table including actions, states, learning rate, greedy rate, discount rate, and so on, is exemplified in 2 two-dimensional potential functions. In the applications of the Q-learning method to two chemical reactions, it is demonstrated that the Q-learning method can predict consistent TS and reaction pathway with those by ab initio calculations. Notably, the PES must be well prepared before using the Q-learning method, and a coarse-to-fine PES scanning scheme is thus introduced to save the computational time while maintaining the accuracy of the Q-learning prediction. This work offers a simple and reliable Q-learning method to search for all possible TS and reaction pathway of a chemical reaction, which may be a new option for effectively exploring the PES in an extensive search manner.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


