{"title":"A case of syndromic congenital hypothyroidism with a 15.2 Mb interstitial deletion on 2q12.3q14.2 involving <i>PAX8</i>.","authors":"Megumi Iwahashi-Odano, Miyuki Kitamura, Satoshi Narumi","doi":"10.1297/cpe.2022-0061","DOIUrl":null,"url":null,"abstract":"<p><p>Paired box 8 (<i>PAX8</i>) mutations are an established genetic cause of congenital hypothyroidism (CH). The majority of these mutations are found in the protein-coding exons of the gene. The proband, a 3-yr-old girl, had tetralogy of Fallot and polydactyly soon after birth. She was diagnosed with CH in the newborn screening for CH. She had a high serum TSH level (239 mU/L) and low free T4 level (0.7 ng/dL). Ultrasonography revealed thyroid hypoplasia. We performed array comparative genomic hybridization because the patient exhibited a variety of symptoms across multiple organ systems. The analysis revealed a novel heterozygous deletion that spanned a 15.2 Mb region in 2q12.3q14.3 (GRCh37; chr2:109,568,260-124,779,449). There were 71 protein-coding genes in this region, including two genes (<i>PAX8</i> and <i>GLI2</i>) associated with congenital endocrine disorders. The common clinical features of the two previously reported patients with a total <i>PAX8</i> deletion and our case were CH, short stature and intellectual disability, but the severity of hypothyroidism and other clinical features were variable. In conclusion, we describe a syndromic CH patient with a novel 2q12.3q14.3 deletion involving <i>PAX8</i>. Patients with CH, whose unifying diagnosis is not obvious, could have a genomic deletion involving <i>PAX8</i>.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":null,"pages":null},"PeriodicalIF":1.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/5b/93/cpe-32-065.PMC9887295.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2022-0061","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
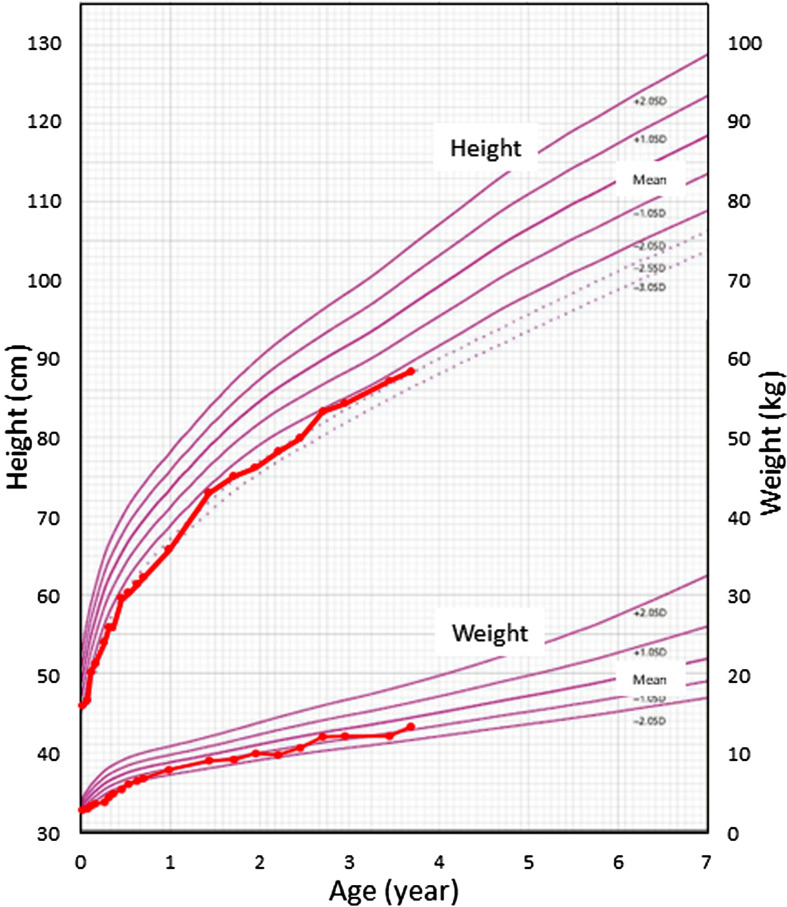
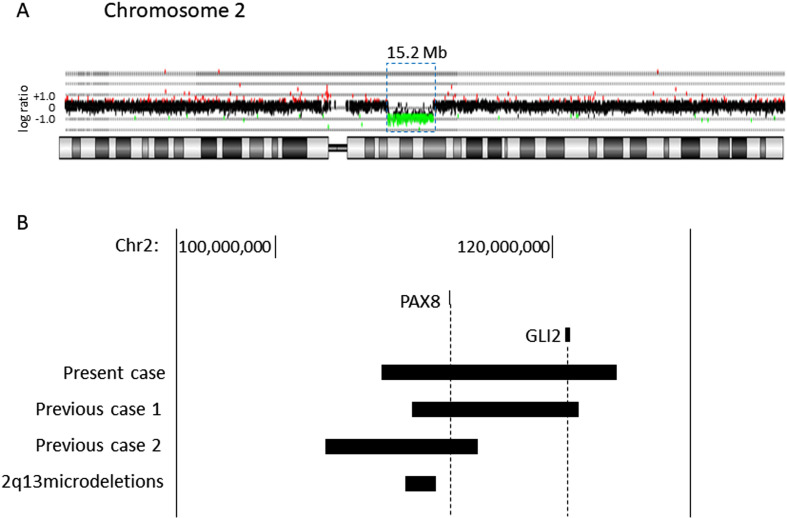
Paired box 8 (PAX8) mutations are an established genetic cause of congenital hypothyroidism (CH). The majority of these mutations are found in the protein-coding exons of the gene. The proband, a 3-yr-old girl, had tetralogy of Fallot and polydactyly soon after birth. She was diagnosed with CH in the newborn screening for CH. She had a high serum TSH level (239 mU/L) and low free T4 level (0.7 ng/dL). Ultrasonography revealed thyroid hypoplasia. We performed array comparative genomic hybridization because the patient exhibited a variety of symptoms across multiple organ systems. The analysis revealed a novel heterozygous deletion that spanned a 15.2 Mb region in 2q12.3q14.3 (GRCh37; chr2:109,568,260-124,779,449). There were 71 protein-coding genes in this region, including two genes (PAX8 and GLI2) associated with congenital endocrine disorders. The common clinical features of the two previously reported patients with a total PAX8 deletion and our case were CH, short stature and intellectual disability, but the severity of hypothyroidism and other clinical features were variable. In conclusion, we describe a syndromic CH patient with a novel 2q12.3q14.3 deletion involving PAX8. Patients with CH, whose unifying diagnosis is not obvious, could have a genomic deletion involving PAX8.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


