[Analysis and identification of suspected snake venom samples using nano-ultra-high performance liquid chromatography-high resolution mass spectrometry].
Abstract
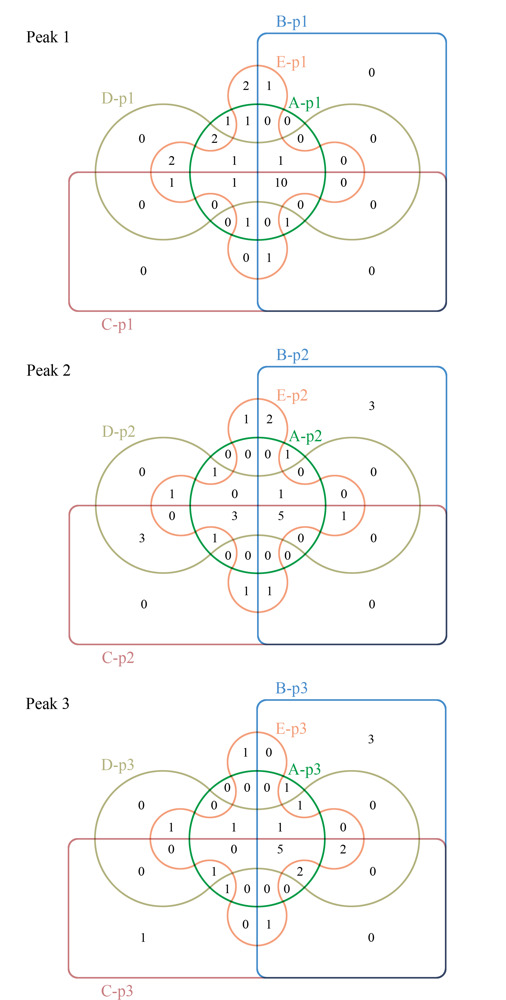
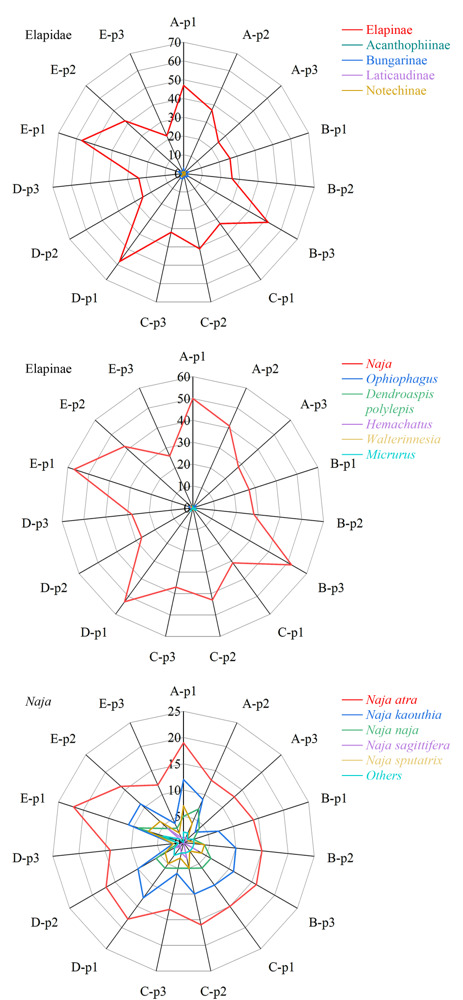
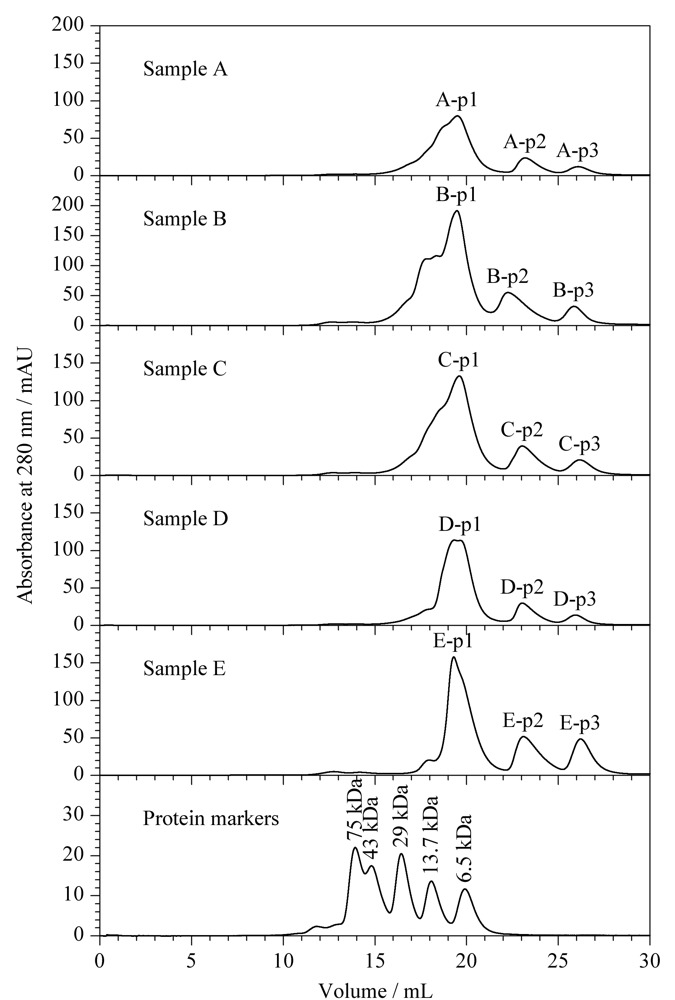
Snake venom is a complex mixture secreted from the glands of poisonous snakes, which contains proteins, peptides, lipids, nucleosides, sugars, amino acids, amines, metal ions, and other components. According to the toxicological classification, snake venoms can be classified as neurotoxins, anticoagulants and procoagulant toxins, cardiac toxins, other toxin proteins, and enzymes. Proteins and peptides are the key components of snake venom. The establishment of rapid, accurate analysis and identification methods for proteins in snake venom is a prerequisite for snake venom-related forensic identification, intoxication events, and pharmaceutical development. Until now, the classical analysis and identification methods have mainly been biochemical or immunoassays for DNA or proteins, such as polymerase chain reaction, agglutination test, enzyme-linked immunosorbent assay, fluorescent immunoassay, and various biosensing approaches. These methods have some limitations such as a high false-positive ratio, low sensitivity, poor anti-interference ability, and limited species discrimination capability. In recent years, with the rapid development of mass spectrometry (MS) techniques, the proteomics of snake venom has also attracted much attention and has contributed to the identification of snake species, in which non-targeted and targeted proteomics represent two main divisions. However, species identification via proteomics is in its infancy in forensic science. First, the tandem MS spectra of peptide sequences are highly complex, which poses a great challenge for the strict and accurate matching of peptides based on the rational speculation of MS fragmentation rules and theoretical calculations in non-targeted proteomics. Second, for the confirmation and identification of unknown substances, reference substances are commonly needed, but those for snake venom are lacking. Proteomics in snake venom identification is still in progress to improve the identification confidence and clarify the identification rules. In this work, a method based on nano-ultra-high performance liquid chromatography-quadrupole-orbitrap high-resolution mass spectrometry (Nano LC-MS/HRMS) and size exclusion chromatography (SEC) was developed for identifying proteins and their source species, with strict rules for five suspected snake venom samples and their contamination in one case. Three SEC elution peaks were obtained from each of the five samples, which were lyophilized and treated with trypsin in solution, and then separated and analyzed by Nano LC-MS/HRMS. First, the Full MS/dd MS2 mode was used for the non-targeted acquisition of peptide information in the samples, and after submission to the Swiss-Prot database, the protein databases of Serpentes, Colubroidea, Elapidae, Elapinae, and Naja were contracted stepwise and compared. A total of 32 proteins from Naja atra were identified under the conditions of both peptide spectrum match and false discovery rate less than 1%, and number of characteristic peptides greater than or equal to two. All of these were derived from ten families of Naja atra, mainly three-finger toxins, metalloproteinases, and phospholipase A2. Proteins D3TTC2, D5LMJ3, Q7T1K6, Q9DEQ3, and Q9YGI4 were the most common among the five samples. Finally, the parallel reaction monitoring mode was adopted to select two unique peptides for each protein for targeted verification. It was considered that a protein in the samples was truly identified when it met the strict standard "the Δm/z of at least 75% y+ and b+ ions of each unique peptide was less than 5 ppm". After these consequently procedures, we identified that all five samples contained the venom of the Naja atra. Our identification method is a systematic and strict example that can provide effective technical support for the forensic identification of snake venom poisoning, as well as for pharmaceutical development toward snake venoms.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


