{"title":"Similarity Index-Probabilistic Confidence Estimation of SARS-CoV-2 Strain Relatedness in Localized Outbreaks.","authors":"Mahmood Y Bilal","doi":"10.3390/epidemiologia3020019","DOIUrl":null,"url":null,"abstract":"<p><p>Outbreaks of SARS-CoV-2 can be attributed to expanding small-scale localized infection subclusters that eventually propagate into regional and global outspread. These infections are driven by spatial as well as temporal mutational dynamics wherein virions diverge genetically as transmission occurs. Mutational similarity or dissimilarity of viral strains, stemming from shared spatiotemporal fields, thence serves as a gauge of relatedness. In our clinical laboratory, molecular epidemiological analyses of strain association are performed qualitatively from genomic sequencing data. These methods however carry a degree of uncertainty when the samples are not qualitatively, with reasonable confidence, deemed identical or dissimilar. We propose a theoretical mathematical model for probability derivation of outbreak-sample similarity as a function of spatial dynamics, shared and different mutations, and total number of samples involved. This Similarity Index utilizes an Essen-Möller ratio of similar and dissimilar mutations between the strains in question. The indices are compared to each strain within an outbreak, and then the final Similarity Index of the outbreak group is calculated to determine quantitative confidence of group relatedness. We anticipate that this model will be useful in evaluating strain associations in SARS-CoV-2 and other viral outbreaks utilizing molecular data.</p>","PeriodicalId":72944,"journal":{"name":"Epidemiolgia (Basel, Switzerland)","volume":"3 2","pages":"238-249"},"PeriodicalIF":0.0000,"publicationDate":"2022-05-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9620881/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epidemiolgia (Basel, Switzerland)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/epidemiologia3020019","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 3
Abstract
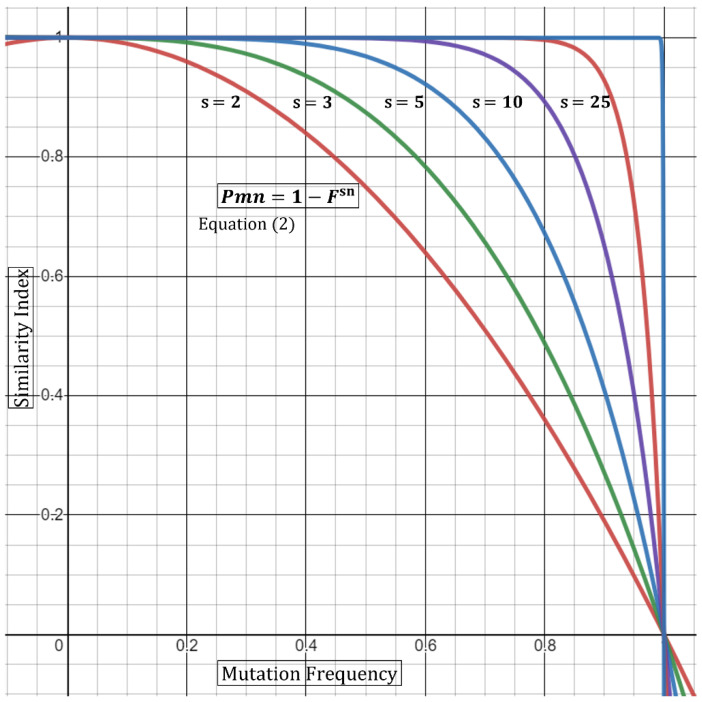
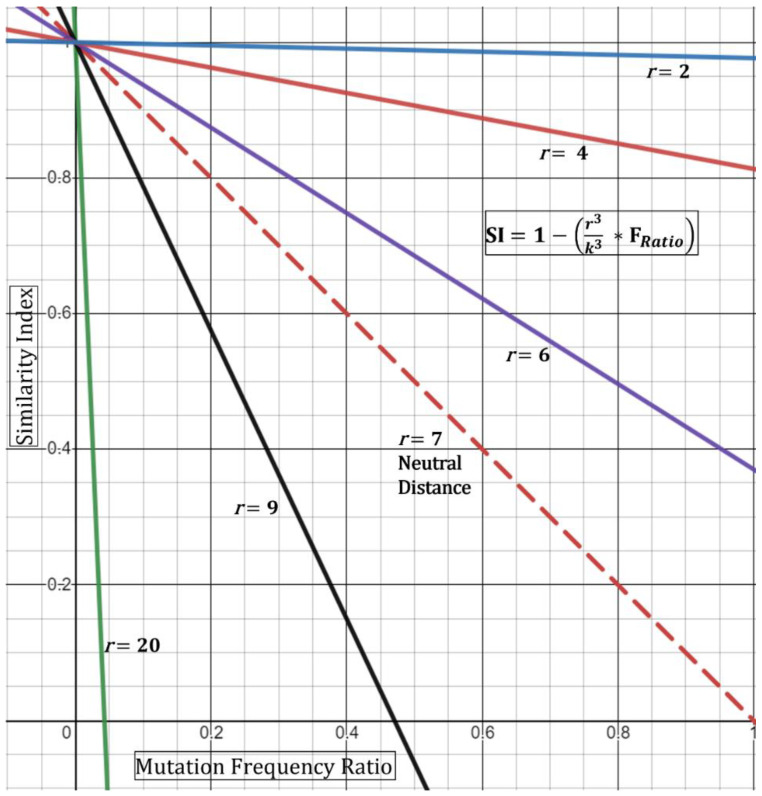
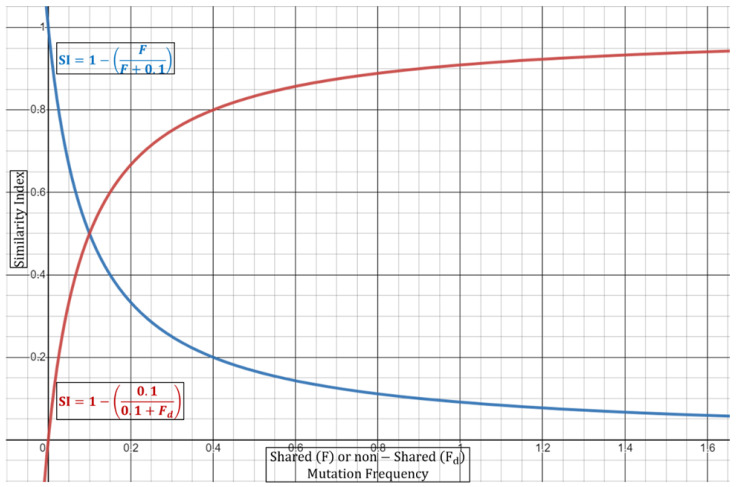
Outbreaks of SARS-CoV-2 can be attributed to expanding small-scale localized infection subclusters that eventually propagate into regional and global outspread. These infections are driven by spatial as well as temporal mutational dynamics wherein virions diverge genetically as transmission occurs. Mutational similarity or dissimilarity of viral strains, stemming from shared spatiotemporal fields, thence serves as a gauge of relatedness. In our clinical laboratory, molecular epidemiological analyses of strain association are performed qualitatively from genomic sequencing data. These methods however carry a degree of uncertainty when the samples are not qualitatively, with reasonable confidence, deemed identical or dissimilar. We propose a theoretical mathematical model for probability derivation of outbreak-sample similarity as a function of spatial dynamics, shared and different mutations, and total number of samples involved. This Similarity Index utilizes an Essen-Möller ratio of similar and dissimilar mutations between the strains in question. The indices are compared to each strain within an outbreak, and then the final Similarity Index of the outbreak group is calculated to determine quantitative confidence of group relatedness. We anticipate that this model will be useful in evaluating strain associations in SARS-CoV-2 and other viral outbreaks utilizing molecular data.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


