A 2-Year-Old Child with Alazami Syndrome with Newly Reported Findings of Immune Deficiency, Periventricular Nodular Heterotopia, and Stroke; Broadening the Phenotype of Alazami.
Kristin D Fauntleroy-Love, Theodore E Wilson, Nurcicek Padem, Meredith R Golomb
{"title":"A 2-Year-Old Child with Alazami Syndrome with Newly Reported Findings of Immune Deficiency, Periventricular Nodular Heterotopia, and Stroke; Broadening the Phenotype of Alazami.","authors":"Kristin D Fauntleroy-Love, Theodore E Wilson, Nurcicek Padem, Meredith R Golomb","doi":"10.1177/2329048X231190784","DOIUrl":null,"url":null,"abstract":"<p><p>Alazami syndrome is a rare autosomal recessive neurodevelopmental disorder due to loss-of-function variants in the La ribonucleoprotein 7 <i>(LARP7)</i> gene. Children with Alazami syndrome are most often affected by a combination of primordial dwarfism, intellectual disability, and distinctive facial features. Previous cases have been primarily found in consanguineous families from the Middle East, Asia, and North Africa. We present a 21-month-old Caucasian male from the Midwest United States with nonconsanguineous parents who presented with frequently reported findings of unusual facial features, poor growth, cardiac and genitourinary findings, and developmental delay; less-frequently reported findings, including transient erythroblastopenia of childhood (TEC) and immune deficiency; and never-before reported findings of periventricular nodular heterotopia and stroke. He developed stroke during a hospitalization for Hemophilus influenzae meningitis. The possible contributions of <i>LARP7</i> to TEC, immune deficiency, brain malformation, and stroke are discussed. Guidelines for the care of Alazami patients are proposed.</p>","PeriodicalId":72572,"journal":{"name":"Child neurology open","volume":"10 ","pages":"2329048X231190784"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/fa/3d/10.1177_2329048X231190784.PMC10388622.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Child neurology open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2329048X231190784","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
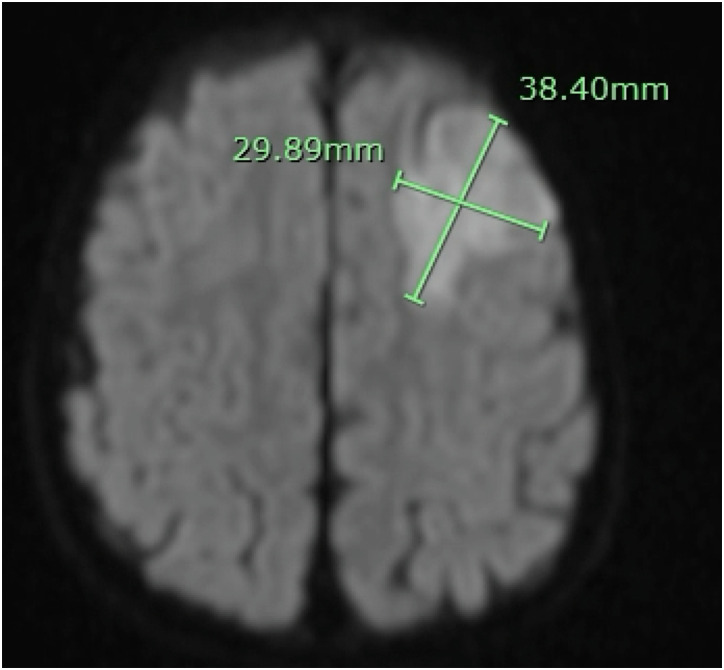
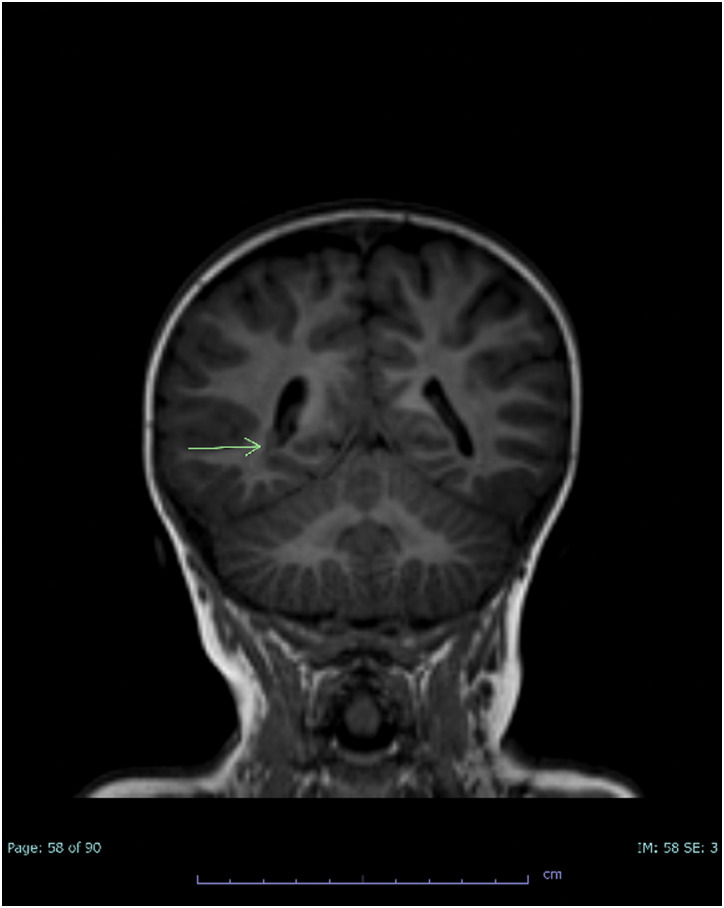
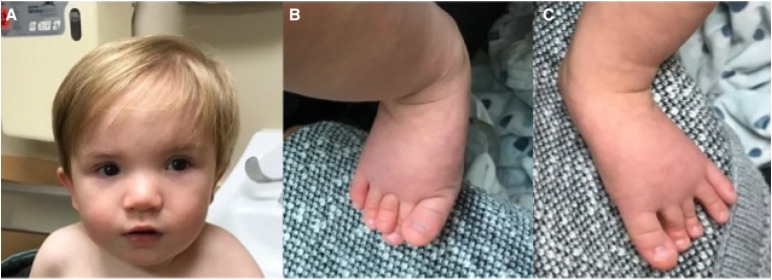
Alazami syndrome is a rare autosomal recessive neurodevelopmental disorder due to loss-of-function variants in the La ribonucleoprotein 7 (LARP7) gene. Children with Alazami syndrome are most often affected by a combination of primordial dwarfism, intellectual disability, and distinctive facial features. Previous cases have been primarily found in consanguineous families from the Middle East, Asia, and North Africa. We present a 21-month-old Caucasian male from the Midwest United States with nonconsanguineous parents who presented with frequently reported findings of unusual facial features, poor growth, cardiac and genitourinary findings, and developmental delay; less-frequently reported findings, including transient erythroblastopenia of childhood (TEC) and immune deficiency; and never-before reported findings of periventricular nodular heterotopia and stroke. He developed stroke during a hospitalization for Hemophilus influenzae meningitis. The possible contributions of LARP7 to TEC, immune deficiency, brain malformation, and stroke are discussed. Guidelines for the care of Alazami patients are proposed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


