Saifur R Khan, Andreea Obersterescu, Erica P Gunderson, Babak Razani, Michael B Wheeler, Brian J Cox
{"title":"metGWAS 1.0: an R workflow for network-driven over-representation analysis between independent metabolomic and meta-genome-wide association studies.","authors":"Saifur R Khan, Andreea Obersterescu, Erica P Gunderson, Babak Razani, Michael B Wheeler, Brian J Cox","doi":"10.1093/bioinformatics/btad523","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>The method of genome-wide association studies (GWAS) and metabolomics combined provide an quantitative approach to pinpoint metabolic pathways and genes linked to specific diseases; however, such analyses require both genomics and metabolomics datasets from the same individuals/samples. In most cases, this approach is not feasible due to high costs, lack of technical infrastructure, unavailability of samples, and other factors. Therefore, an unmet need exists for a bioinformatics tool that can identify gene loci-associated polymorphic variants for metabolite alterations seen in disease states using standalone metabolomics.</p><p><strong>Results: </strong>Here, we developed a bioinformatics tool, metGWAS 1.0, that integrates independent GWAS data from the GWAS database and standalone metabolomics data using a network-based systems biology approach to identify novel disease/trait-specific metabolite-gene associations. The tool was evaluated using standalone metabolomics datasets extracted from two metabolomics-GWAS case studies. It discovered both the observed and novel gene loci with known single nucleotide polymorphisms when compared to the original studies.</p><p><strong>Availability and implementation: </strong>The developed metGWAS 1.0 framework is implemented in an R pipeline and available at: https://github.com/saifurbd28/metGWAS-1.0.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491949/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad523","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Motivation: The method of genome-wide association studies (GWAS) and metabolomics combined provide an quantitative approach to pinpoint metabolic pathways and genes linked to specific diseases; however, such analyses require both genomics and metabolomics datasets from the same individuals/samples. In most cases, this approach is not feasible due to high costs, lack of technical infrastructure, unavailability of samples, and other factors. Therefore, an unmet need exists for a bioinformatics tool that can identify gene loci-associated polymorphic variants for metabolite alterations seen in disease states using standalone metabolomics.
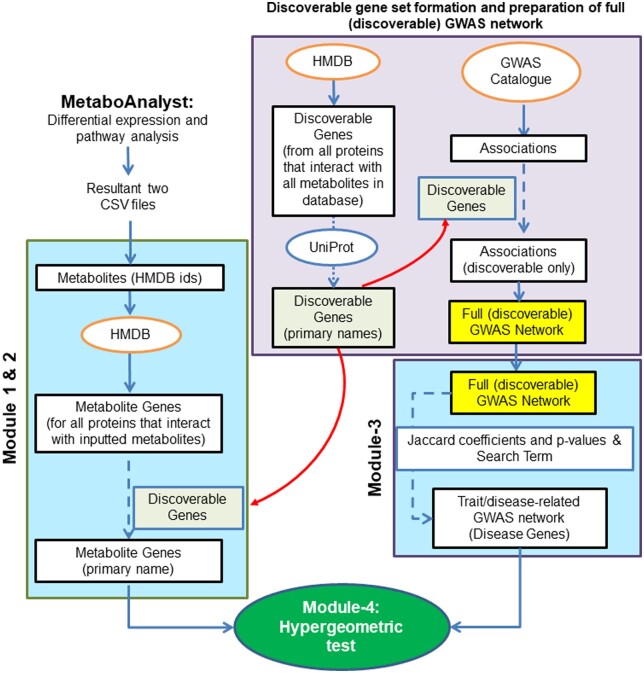
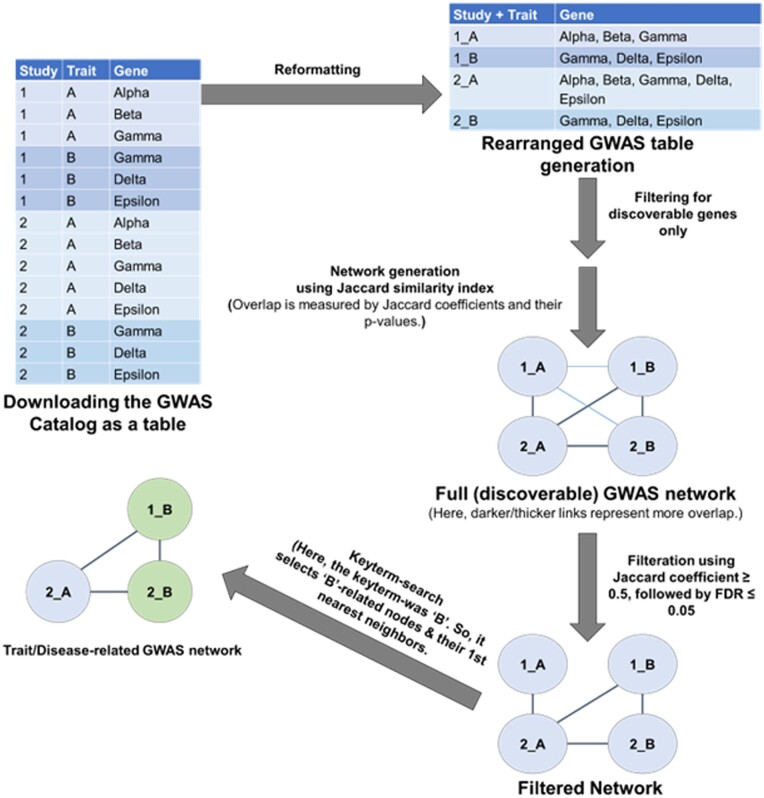
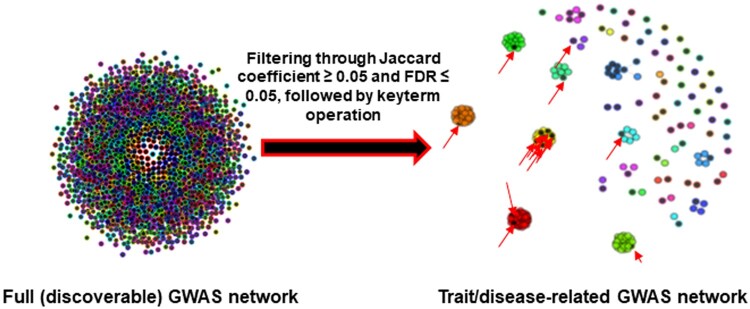
Results: Here, we developed a bioinformatics tool, metGWAS 1.0, that integrates independent GWAS data from the GWAS database and standalone metabolomics data using a network-based systems biology approach to identify novel disease/trait-specific metabolite-gene associations. The tool was evaluated using standalone metabolomics datasets extracted from two metabolomics-GWAS case studies. It discovered both the observed and novel gene loci with known single nucleotide polymorphisms when compared to the original studies.
Availability and implementation: The developed metGWAS 1.0 framework is implemented in an R pipeline and available at: https://github.com/saifurbd28/metGWAS-1.0.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


