David O Oladejo, Gbolahan O Duselu, Titilope M Dokunmu, Itunuoluwa Isewon, Jelili Oyelade, Esther Okafor, Emeka Ej Iweala, Ezekiel Adebiyi
{"title":"<i>In silico</i> Structure Prediction, Molecular Docking, and Dynamic Simulation of <i>Plasmodium falciparum</i> AP2-I Transcription Factor.","authors":"David O Oladejo, Gbolahan O Duselu, Titilope M Dokunmu, Itunuoluwa Isewon, Jelili Oyelade, Esther Okafor, Emeka Ej Iweala, Ezekiel Adebiyi","doi":"10.1177/11779322221149616","DOIUrl":null,"url":null,"abstract":"<p><p><i>Plasmodium falciparum</i> Apicomplexan Apetala 2 Invasion (<i>Pf</i>AP2-I) transcription factor (TF) is a protein that regulates the expression of a subset of gene families involved in <i>P. falciparum</i> red blood cell (RBC) invasion. Inhibiting <i>Pf</i>AP2-I TF with small molecules represents a potential new antimalarial therapeutic target to combat drug resistance, which this study aims to achieve. The 3D model structure of <i>Pf</i>AP2-I was predicted <i>ab initio</i> using ROBETTA prediction tool and was validated using Save server 6.0 and MolProbity. Computed Atlas of Surface Topography of proteins (CASTp) 3.0 was used to predict the active sites of the <i>Pf</i>AP2-I modeled structure. Pharmacophore modeling of the control ligand and <i>Pf</i>AP2-I modeled structure was carried out using the Pharmit server to obtain several compounds used for molecular docking analysis. Molecular docking and postdocking studies were conducted using AutoDock vina and Discovery studio. The designed ligands' toxicity predictions and <i>in silico</i> drug-likeness were performed using the SwissADME predictor and OSIRIS Property Explorer. The modeled protein structure from the ROBETTA showed a validation result of 96.827 for ERRAT, 90.2% of the amino acid residues in the most favored region for the Ramachandran plot, and MolProbity score of 1.30 in the 98th percentile. Five (5) best hit compounds from molecular docking analysis were selected based on their binding affinity (between -8.9 and -11.7 Kcal/mol) to the active site of <i>Pf</i>AP2-I and were considered for postdocking studies. For the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties, compound MCULE-7146940834 had the highest drug score (0.63) and drug-likeness (6.76). MCULE-7146940834 maintained a stable conformation within the flexible protein's active site during simulation. The good, estimated binding energies, drug-likeness, drug score, and molecular dynamics simulation interaction observed for MCULE-7146940834 against <i>Pf</i>AP2-I show that MCULE-7146940834 can be considered a lead candidate for <i>Pf</i>AP2-I inhibition. Experimental validations should be carried out to ascertain the efficacy of these predicted best hit compounds.</p>","PeriodicalId":9065,"journal":{"name":"Bioinformatics and Biology Insights","volume":"17 ","pages":"11779322221149616"},"PeriodicalIF":2.3000,"publicationDate":"2023-01-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/14/a7/10.1177_11779322221149616.PMC9871981.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics and Biology Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11779322221149616","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
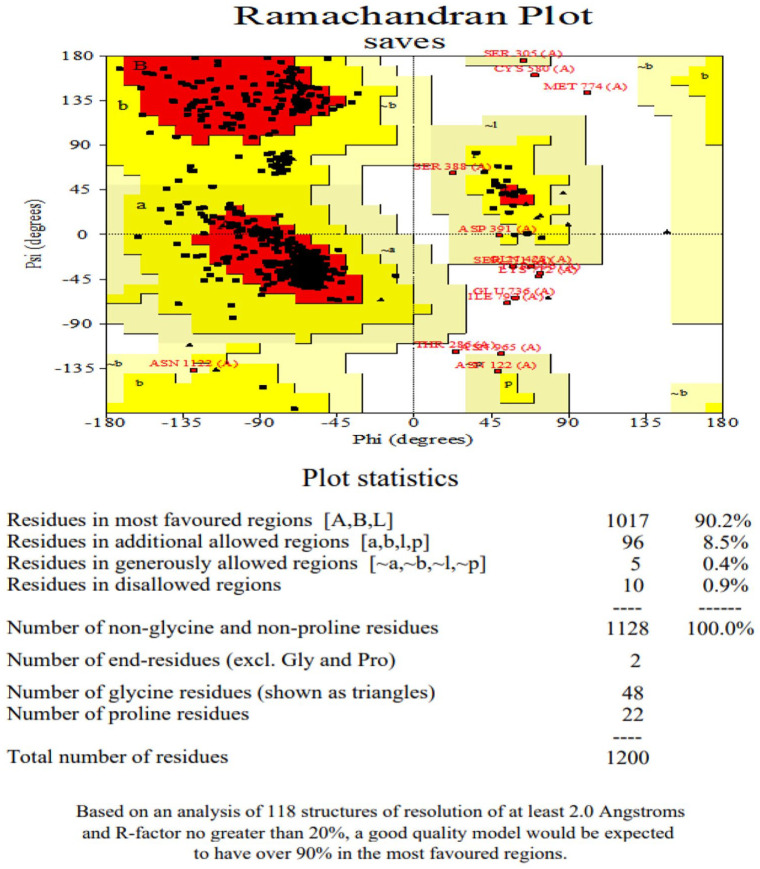
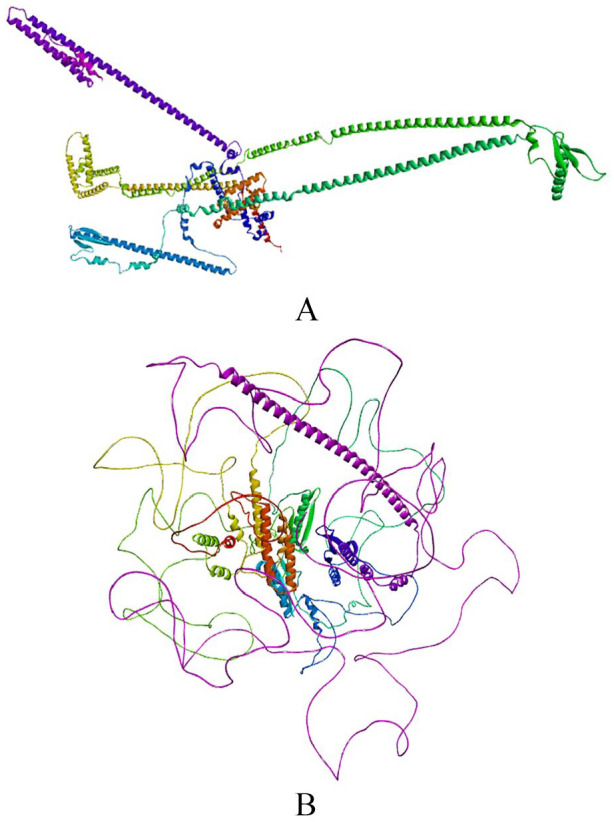
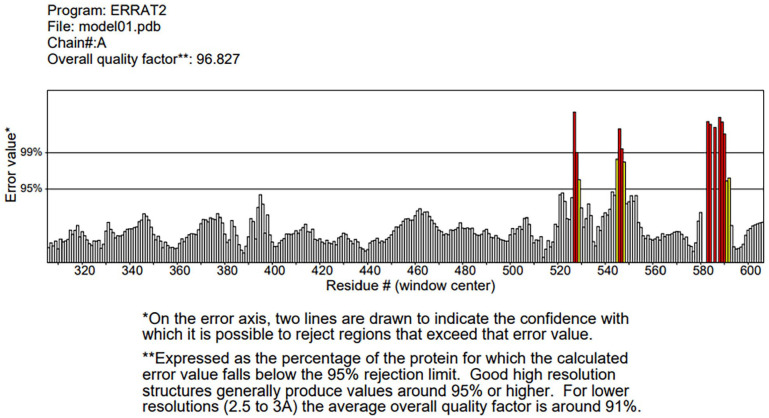
Plasmodium falciparum Apicomplexan Apetala 2 Invasion (PfAP2-I) transcription factor (TF) is a protein that regulates the expression of a subset of gene families involved in P. falciparum red blood cell (RBC) invasion. Inhibiting PfAP2-I TF with small molecules represents a potential new antimalarial therapeutic target to combat drug resistance, which this study aims to achieve. The 3D model structure of PfAP2-I was predicted ab initio using ROBETTA prediction tool and was validated using Save server 6.0 and MolProbity. Computed Atlas of Surface Topography of proteins (CASTp) 3.0 was used to predict the active sites of the PfAP2-I modeled structure. Pharmacophore modeling of the control ligand and PfAP2-I modeled structure was carried out using the Pharmit server to obtain several compounds used for molecular docking analysis. Molecular docking and postdocking studies were conducted using AutoDock vina and Discovery studio. The designed ligands' toxicity predictions and in silico drug-likeness were performed using the SwissADME predictor and OSIRIS Property Explorer. The modeled protein structure from the ROBETTA showed a validation result of 96.827 for ERRAT, 90.2% of the amino acid residues in the most favored region for the Ramachandran plot, and MolProbity score of 1.30 in the 98th percentile. Five (5) best hit compounds from molecular docking analysis were selected based on their binding affinity (between -8.9 and -11.7 Kcal/mol) to the active site of PfAP2-I and were considered for postdocking studies. For the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties, compound MCULE-7146940834 had the highest drug score (0.63) and drug-likeness (6.76). MCULE-7146940834 maintained a stable conformation within the flexible protein's active site during simulation. The good, estimated binding energies, drug-likeness, drug score, and molecular dynamics simulation interaction observed for MCULE-7146940834 against PfAP2-I show that MCULE-7146940834 can be considered a lead candidate for PfAP2-I inhibition. Experimental validations should be carried out to ascertain the efficacy of these predicted best hit compounds.
期刊介绍:
Bioinformatics and Biology Insights is an open access, peer-reviewed journal that considers articles on bioinformatics methods and their applications which must pertain to biological insights. All papers should be easily amenable to biologists and as such help bridge the gap between theories and applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


