{"title":"Hemolytic Anemia Requiring Splenectomy in Leigh-Like Syndrome due to the Variant m.10191T>C in <i>MT</i>-<i>ND3</i>.","authors":"Shaundra M Newstead, Josef Finsterer","doi":"10.14740/jh1122","DOIUrl":null,"url":null,"abstract":"Leigh syndrome is a syndromic mitochondrial disorder (MID), most commonly and clinically characterized by early-onset cognitive impairment, developmental delay, seizures, hypotonia, nutritional problems, and symmetric changes in the basal ganglia and brainstem. The effects of organs other than the brain, such as the heart, intestines, endocrine system, or blood cells, have only rarely been reported. Leigh syndrome is mainly congenital in children and rarely occurs in adults. Hemolytic anemias are a heterogeneous group of hematologic disorders in which red blood cells (RBCs) are destroyed in either an extravascular or intravascular manner [1]. One cause of hemolytic anemia is poikilocytosis, which describes the state of the RBC bimembranes no longer being a biconcave disc shape, but instead, any type of shape [2]. When this occurs, phosphatidylserine from the inner membrane is externally exposed, and the blood cell is marked for destruction by the complement system or sequestered by splenic macrophages [3]. This case of a patient with Leigh-like syndrome (LLS) describes a mostly extravascular acquired hemolytic anemia and cytopenia, due to splenomegalic hypersplenism secondary to poikilocytosis, which partly resolved post-splenectomy. The patient is a 32-year-old Caucasian female, previously described [4] to have LLS due to the variant m.10191T>C in MT-ND3. The mutation was detected in buccal mucosa cells. Heteroplasmy was not determined as it was not covered by insurance. The patient presented with anemia at the age of 25, in August 2015, when blood counts revealed a decreased hemoglobin (Hb) and hematocrit (HCT), with elevated erythrocyte sedimentation rate (ESR) (Table 1). However, complete blood count (CBC) from age 14 already showed Hb of 11 12 g/ dL. In September 2015, the RBC, Hb, and HCT were low, and red cell distribution width standard deviation (RDW-SD) was elevated (Supplementary Material 1, www.thejh.org). Polyspecific direct Coombs antibody test was negative. Bone marrow biopsy (BMB) revealed 100% hypercellular marrow, erythroid hyperplasia and increased reticulin fibers. Despite the negative antibody test, the patient was placed on cyclosporine A (100 mg/day) for a presumptive autoimmune process. In October 2015, haptoglobin was low. Paroxysmal nocturnal hemoglobinuria testing was negative and computed tomography (CT) revealed splenomegaly (Supplementary Material 1, www.thejh.org). In January 2016, the RBC, HCT and Hb were lower compared to previous results. Coombs antibody was checked again and still negative, while lactic dehydrogenase was low for hemolysis. The patient received blood transfusion. In February 2016, Hb, RBC, HCT, platelet (PLT) and WBC were all at their lowest points. Activated partial thromboplastin time was prolonged at 37 s (normal: 22 31 s). Another blood transfusion was given, a diagnosis of cytopenia secondary to hypersplenism was made, and the patient was taken off cyclosporine. ESR was extremely elevated. Schistocytes and other RBC morphology were negative. Glucose-6-phosphate dehydrogenase (G6PD) levels measured after transfusion were normal. Serum protein electrophoresis was normal except for low alpha-2. Immunoglobulins G, A and M, rheumatoid factor, antinuclear antibodies, and complements C3 and C4 were normal. BMB revealed 100% hypercellular marrow, erythroid hyperplasia, normal iron stores and no lymphoid aggregates or reticulin fibers. The splenectomy was performed in May 2016. This choice was made because other diseases had been ruled out, along with poikilocytosis and the spleen’s continued growth with a trend toward pancytopenia. The size on CT was larger than normal gross pathological examination, which revealed an enlarged spleen weighing 404 g (normal female: 50 250 g), and spleen pathology revealed extramedullary hematopoiesis, congestion of the red pulp, scattered macrophages and follicular hyperplasia. The pathologist determined that the findings were consistent with acquired hemolytic anemia. The patient experienced severe thrombocytosis of 1,600 × 103/μL (normal: 150 450 × 103/μL), but examinations for thrombosis were negative (Supplementary Material 1, www.thejh.org). In September 2016, haptoglobin was elevated, RBC, Hb and HCT had improved, WBC had fully recovered, and thrombocytosis had lowered. The RDW-SD had increased even higher than the peak of hemolysis in February 2016. Additionally, RBC morphology revealed 3+ anisocytosis, 3+ macrocytosis, +1 elliptocytosis and +1 schistocytosis (Fig. 1). A final BMB surprisingly still revealed a 100% hypercellular marrow. Reticulin was normal and iron storage was depleted. Lymphoid Manuscript submitted April 4, 2023, accepted May 12, 2023 Published online July 12, 2023","PeriodicalId":15964,"journal":{"name":"Journal of hematology","volume":"12 4","pages":"197-200"},"PeriodicalIF":1.3000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ae/61/jh-12-197.PMC10482609.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14740/jh1122","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
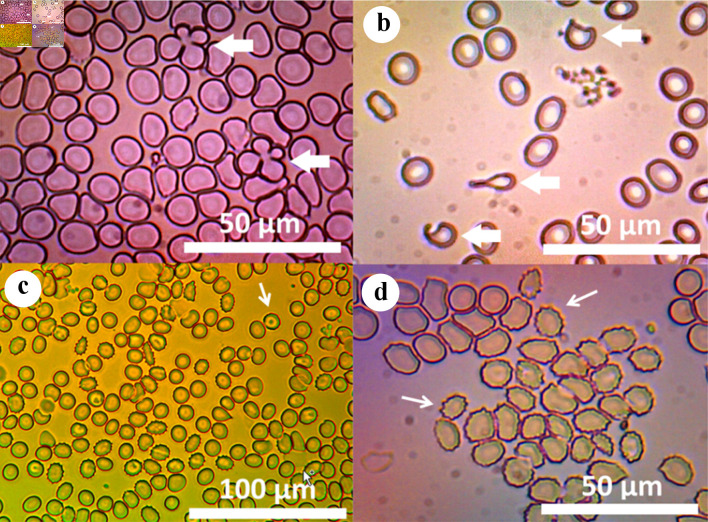
Leigh syndrome is a syndromic mitochondrial disorder (MID), most commonly and clinically characterized by early-onset cognitive impairment, developmental delay, seizures, hypotonia, nutritional problems, and symmetric changes in the basal ganglia and brainstem. The effects of organs other than the brain, such as the heart, intestines, endocrine system, or blood cells, have only rarely been reported. Leigh syndrome is mainly congenital in children and rarely occurs in adults. Hemolytic anemias are a heterogeneous group of hematologic disorders in which red blood cells (RBCs) are destroyed in either an extravascular or intravascular manner [1]. One cause of hemolytic anemia is poikilocytosis, which describes the state of the RBC bimembranes no longer being a biconcave disc shape, but instead, any type of shape [2]. When this occurs, phosphatidylserine from the inner membrane is externally exposed, and the blood cell is marked for destruction by the complement system or sequestered by splenic macrophages [3]. This case of a patient with Leigh-like syndrome (LLS) describes a mostly extravascular acquired hemolytic anemia and cytopenia, due to splenomegalic hypersplenism secondary to poikilocytosis, which partly resolved post-splenectomy. The patient is a 32-year-old Caucasian female, previously described [4] to have LLS due to the variant m.10191T>C in MT-ND3. The mutation was detected in buccal mucosa cells. Heteroplasmy was not determined as it was not covered by insurance. The patient presented with anemia at the age of 25, in August 2015, when blood counts revealed a decreased hemoglobin (Hb) and hematocrit (HCT), with elevated erythrocyte sedimentation rate (ESR) (Table 1). However, complete blood count (CBC) from age 14 already showed Hb of 11 12 g/ dL. In September 2015, the RBC, Hb, and HCT were low, and red cell distribution width standard deviation (RDW-SD) was elevated (Supplementary Material 1, www.thejh.org). Polyspecific direct Coombs antibody test was negative. Bone marrow biopsy (BMB) revealed 100% hypercellular marrow, erythroid hyperplasia and increased reticulin fibers. Despite the negative antibody test, the patient was placed on cyclosporine A (100 mg/day) for a presumptive autoimmune process. In October 2015, haptoglobin was low. Paroxysmal nocturnal hemoglobinuria testing was negative and computed tomography (CT) revealed splenomegaly (Supplementary Material 1, www.thejh.org). In January 2016, the RBC, HCT and Hb were lower compared to previous results. Coombs antibody was checked again and still negative, while lactic dehydrogenase was low for hemolysis. The patient received blood transfusion. In February 2016, Hb, RBC, HCT, platelet (PLT) and WBC were all at their lowest points. Activated partial thromboplastin time was prolonged at 37 s (normal: 22 31 s). Another blood transfusion was given, a diagnosis of cytopenia secondary to hypersplenism was made, and the patient was taken off cyclosporine. ESR was extremely elevated. Schistocytes and other RBC morphology were negative. Glucose-6-phosphate dehydrogenase (G6PD) levels measured after transfusion were normal. Serum protein electrophoresis was normal except for low alpha-2. Immunoglobulins G, A and M, rheumatoid factor, antinuclear antibodies, and complements C3 and C4 were normal. BMB revealed 100% hypercellular marrow, erythroid hyperplasia, normal iron stores and no lymphoid aggregates or reticulin fibers. The splenectomy was performed in May 2016. This choice was made because other diseases had been ruled out, along with poikilocytosis and the spleen’s continued growth with a trend toward pancytopenia. The size on CT was larger than normal gross pathological examination, which revealed an enlarged spleen weighing 404 g (normal female: 50 250 g), and spleen pathology revealed extramedullary hematopoiesis, congestion of the red pulp, scattered macrophages and follicular hyperplasia. The pathologist determined that the findings were consistent with acquired hemolytic anemia. The patient experienced severe thrombocytosis of 1,600 × 103/μL (normal: 150 450 × 103/μL), but examinations for thrombosis were negative (Supplementary Material 1, www.thejh.org). In September 2016, haptoglobin was elevated, RBC, Hb and HCT had improved, WBC had fully recovered, and thrombocytosis had lowered. The RDW-SD had increased even higher than the peak of hemolysis in February 2016. Additionally, RBC morphology revealed 3+ anisocytosis, 3+ macrocytosis, +1 elliptocytosis and +1 schistocytosis (Fig. 1). A final BMB surprisingly still revealed a 100% hypercellular marrow. Reticulin was normal and iron storage was depleted. Lymphoid Manuscript submitted April 4, 2023, accepted May 12, 2023 Published online July 12, 2023

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


